Clear Sky Science · pt
Usando as referências lineares do pangenoma para descobrir variantes de autismo perdidas
Por que alterações ocultas no DNA importam para o autismo
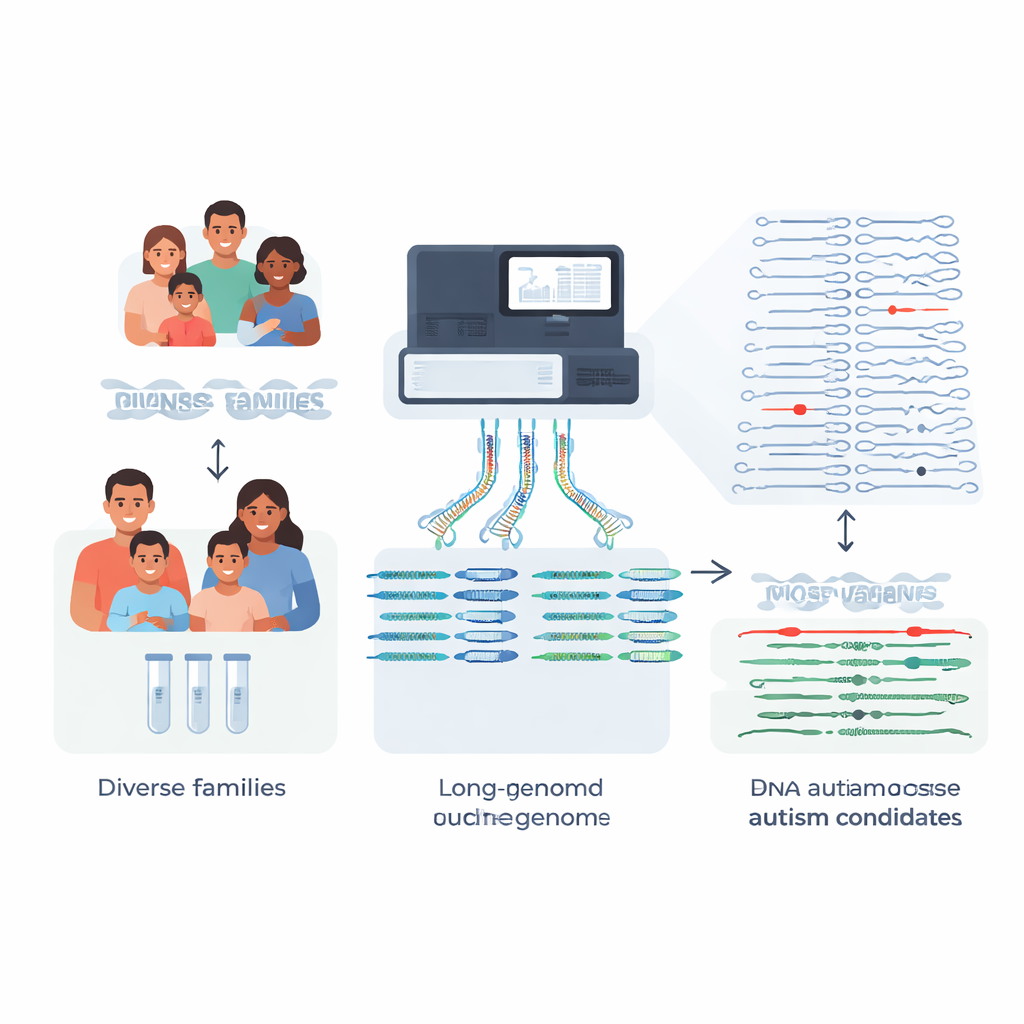
A maioria das famílias que recorrem a testes genéticos para uma criança autista espera respostas claras, mas cerca de quatro em cada cinco não recebem uma explicação genética definitiva. Este estudo aborda uma razão importante para isso: muitas alterações no DNA com impacto são complexas demais para testes padrão detectarem. Ao construir genomas quase completos de 189 pessoas de 51 famílias afetadas pelo autismo e compará-los a uma nova referência mais rica — o “pangenoma” — os pesquisadores mostram como o sequenciamento avançado pode revelar mutações raras e previamente invisíveis que podem ajudar a explicar alguns casos de autismo e condições relacionadas.
Olhando além dos testes genéticos padrão
Os testes clínicos tradicionais dependem de trechos curtos de DNA para escanear o genoma de uma pessoa. Isso funciona bem para muitas mudanças de uma única letra, mas frequentemente falha em regiões repetitivas ou estruturalmente complexas — exatamente onde algumas mutações de grande efeito se escondem. A equipe focou em famílias nas quais testes anteriores com leituras curtas do genoma, exoma ou painéis de genes não haviam encontrado uma causa para sintomas de autismo ou semelhantes aos da síndrome de Rett. Usando sequenciamento de leitura longa, que lê trechos muito maiores de DNA, eles construíram montagens genômicas de alta qualidade e com fase para 189 indivíduos. Isso significa que puderam reconstruir as duas cópias de cada cromossomo de cada pessoa, uma herdada de cada progenitor, com pouquíssimas lacunas.
Variantes estruturais: mudanças grandes com grandes efeitos
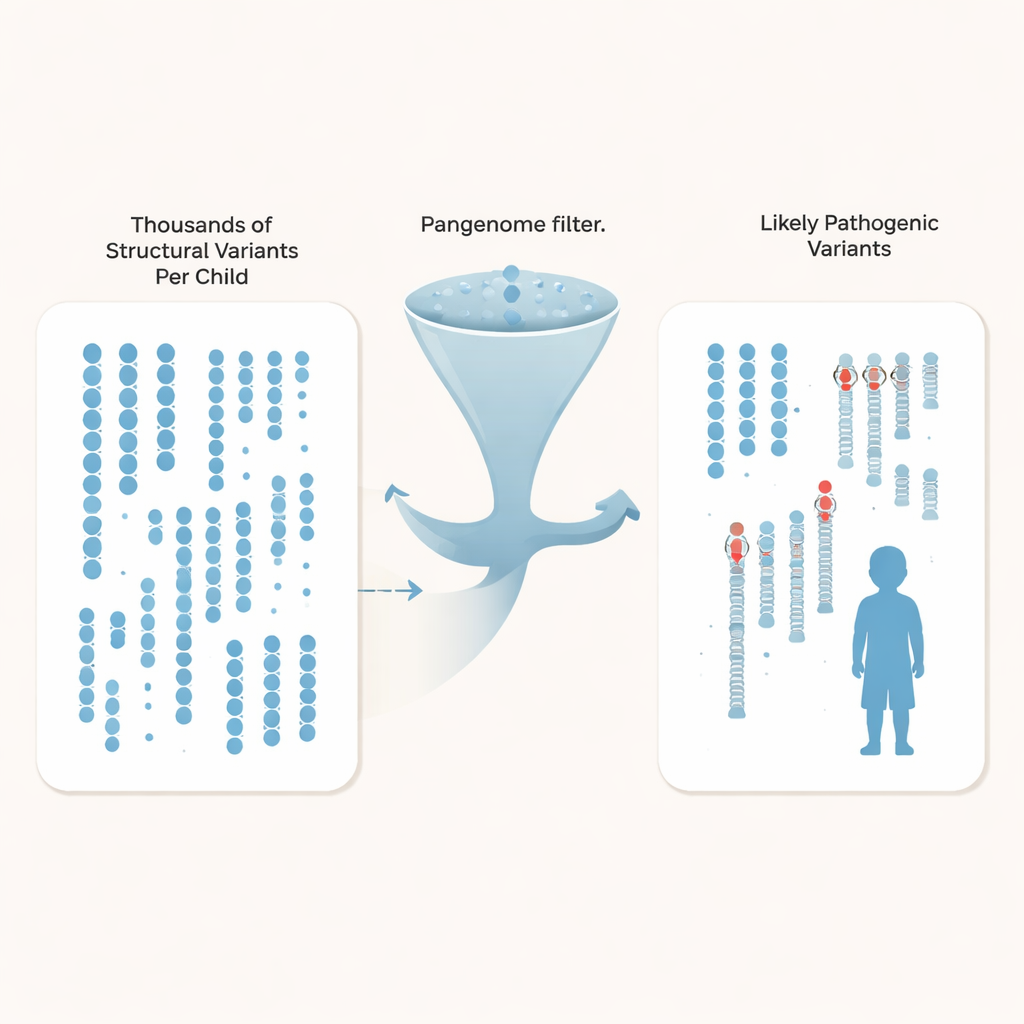
Em vez de apenas rastrear diferenças de uma letra, os pesquisadores se concentraram em variantes estruturais — inserções, deleções e rearranjos que afetam pelo menos 50 bases de DNA e podem interromper genes ou seus interruptores regulatórios. Cada criança carregava cerca de 27.000 dessas variantes, mas a grande maioria são diferenças de fundo inofensivas compartilhadas pela população. Comparando suas famílias com centenas de genomas de controle profundamente sequenciados do pangenoma, provenientes de diversas ancestrias, a equipe conseguiu filtrar mais de 97% das variantes estruturais comuns para cada criança, restando cerca de 600 candidatos raros por genoma, e tão poucos quanto cerca de 200 quando se usa o maior conjunto de controle.
Encontrando mutações perdidas em genes de risco conhecidos
Com o espaço de busca drasticamente reduzido, os autores integraram várias linhas de evidência: genes já associados ao autismo e a transtornos do neurodesenvolvimento, regiões regulatórias ativas no córtex humano em desenvolvimento e padrões de herança dentro de cada família. Eles descobriram três mutações claramente patogênicas que testes anteriores haviam perdido. Isso incluiu um novo sinal de parada no gene SYNGAP1, importante para a função sináptica, e uma deleção que remove o último éxon de MECP2, um gene chave da síndrome de Rett, mesmo que o paciente já tivesse passado por múltiplos testes clínicos prévios. Também confirmaram uma alteração causadora de doença em TBL1XR1, um gene que interage com MECP2. No total, destacaram nove variantes estruturais adicionais — frequentemente herdadas e localizadas em regiões regulatórias próximas a genes relacionados ao cérebro — como fortes candidatas para testes funcionais futuros.
O que o estudo não encontrou — e por que isso ainda é importante
Apesar dessa busca profunda, os autores não observaram um excesso claro e geral de variantes estruturais em crianças autistas em comparação com seus irmãos não afetados, pelo menos neste tamanho de amostra modesto. Houve, no entanto, um indício de mais alterações estruturais no cromossomo X em meninas afetadas, e as montagens quase completas dos cromossomos X e Y permitiram detectar padrões incomuns, como um forte enviesamento na inativação do cromossomo X. Essas características podem se tornar pistas importantes à medida que mais famílias forem estudadas. De forma crucial, o trabalho demonstra que o sequenciamento de leitura longa pode recuperar variantes patogênicas que métodos de leitura curta deixam escapar, especialmente em partes difíceis do genoma e nas regiões de controle que ajustam a atividade gênica.
O que isso significa para famílias e para testes futuros
Para as famílias, o impacto imediato é modesto, mas significativo: entre esses casos difíceis de resolver, cerca de 6% receberam um diagnóstico genético claro, e quase uma em cada cinco obteve novos candidatos fortes para investigação. Para a área, a mensagem é maior. À medida que referências genômicas mais diversas e completas forem adicionadas ao pangenoma e o sequenciamento de leitura longa se tornar mais acessível, os clínicos poderão descartar rapidamente mudanças estruturais comuns e concentrar-se em um pequeno conjunto de variantes raras e potencialmente deletérias em cada paciente. Essa mudança pode, gradualmente, transformar muitos dos atuais casos “não solucionados” de autismo em casos nos quais a biologia subjacente — e possíveis caminhos para suporte e tratamento — sejam muito melhor compreendidos.
Citação: Sui, Y., Lin, J., Noyes, M.D. et al. Using the linear references from the pangenome to discover missing autism variants. Nat Commun 17, 1681 (2026). https://doi.org/10.1038/s41467-026-68378-4
Palavras-chave: genética do autismo, sequenciamento de leitura longa, variantes estruturais, pangenoma humano, síndrome de Rett