Clear Sky Science · pt
Melhores práticas e ferramentas em R e Python para processamento estatístico e visualização de dados de lipidômica e metabolômica
Por que transformar números de laboratório em imagens claras é importante
Instrumentos modernos conseguem medir milhares de pequenas moléculas—lipídios e outros metabólitos—em uma única gota de sangue ou tecido. Essas medições contêm pistas sobre riscos de doença, respostas a tratamentos e como nossos corpos reagem à dieta ou ao envelhecimento. Mas o resultado bruto não é uma resposta pronta: é uma tabela enorme de números que precisa ser limpa, analisada e transformada em imagens compreensíveis. Este artigo explica como pesquisadores podem usar duas linguagens de programação populares, R e Python, para fazer isso de forma confiável, transparente e com gráficos de qualidade para publicação.
Das medições químicas a tabelas de dados complexas
Em lipidômica e metabolômica, espectrometria de massa e cromatografia geram grandes conjuntos de dados em que cada linha é uma amostra e cada coluna é uma molécula. Essas tabelas raramente se comportam como exemplos didáticos organizados. Contêm valores ausentes, outliers e distribuições assimétricas em que poucas moléculas exibem níveis extremamente altos. As concentrações podem abranger várias ordens de grandeza e ser influenciadas por idade, sexo, dieta, medicamentos, ritmos diários e problemas técnicos como deriva do equipamento ou efeitos de lote. Grupos internacionais de especialistas publicaram diretrizes para padronizar coleta, processamento e relato de amostras, mas mesmo com boas práticas laboratoriais, um processamento estatístico cuidadoso continua essencial para extrair sinais biológicos verdadeiros desse fundo ruidoso.
Limpeza e preparação dos números
Antes que qualquer comparação entre grupos saudáveis e doentes faça sentido, os dados precisam ser preparados. A revisão descreve como surgem os valores ausentes—por falhas aleatórias, limitações do instrumento ou interferência do sinal—e explica quando eles podem ser ignorados em segurança, quando devem ser re-mesurados e como podem ser estimados sensatamente (imputados) usando métodos como k-vizinhos mais próximos, florestas aleatórias ou substituição por valores baixos simples. Em seguida, os autores delineiam estratégias de normalização que reduzem variação indesejada, por exemplo corrigindo efeitos de lote com amostras de controle de qualidade ou ajustando diferenças na quantidade de amostra. Depois, discutem transformações como logaritmos—que domam caudas longas à direita nos dados—e métodos de escalonamento que colocam todas as moléculas em um pé de igualdade para que compostos altamente variáveis não dominem as análises posteriores. 
Testes estatísticos e narrativas visuais
Com os dados adequadamente preparados, uma gama de ferramentas estatísticas entra em ação. Para moléculas individuais, pesquisadores podem calcular razões de mudança (fold changes) e usar testes clássicos como o teste t ou seus equivalentes não paramétricos (como o teste de Mann–Whitney) para verificar se os níveis diferem entre grupos. Para comparações envolvendo vários grupos, são apresentadas metodologias como ANOVA ou o teste de Kruskal–Wallis, acompanhadas por procedimentos post hoc para apontar quais grupos diferem. O poder desses testes se revela quando seus resultados são visualizados de forma clara. O artigo destaca box plots (incluindo versões melhoradas para dados assimétricos), violin plots e volcano plots que combinam tamanho de efeito e significância estatística. Para lipídios, descrevem-se visuais mais especializados, como redes lipídicas que mostram alterações coordenadas em classes inteiras, e gráficos de cadeias acilas graxas que revelam padrões no comprimento da cadeia de carbono e grau de saturação.
Ver padrões em muitas variáveis ao mesmo tempo
Como cada amostra pode ter centenas ou milhares de moléculas medidas, métodos multivariados são cruciais. A revisão explica como a análise de componentes principais (PCA) comprime essa complexidade em alguns novos eixos que capturam as principais direções de variação, permitindo checagens rápidas de separação de grupos, efeitos de lote ou estabilidade analítica. Métodos não lineares mais avançados, incluindo t-SNE e UMAP, podem revelar aglomerados e estruturas sutis em espaços de alta dimensão. Para situações em que o objetivo é classificar amostras—por exemplo, distinguir pacientes de controles—os autores descrevem abordagens supervisionadas baseadas em Partial Least Squares e sua extensão ortogonal (PLS-DA e OPLS-DA). Esses métodos vinculam perfis moleculares às etiquetas das amostras, dão suporte à seleção de características e costumam ser resumidos com gráficos de scores, gráficos de loadings e curvas ROC (receiver operating characteristic).
Conjuntos de ferramentas práticos em R e Python
Para ajudar iniciantes a passar da teoria à prática, o artigo faz um panorama do amplo ecossistema de pacotes de software. Em R, coleções como tidyverse e tidymodels simplificam o tratamento e modelagem de dados, enquanto ggplot2 e pacotes complementares como ggpubr, ggstatsplot e tidyplots facilitam a geração de figuras prontas para publicação. Bibliotecas especializadas lidam com PCA, clustering e modelos baseados em PLS, e pacotes do Bioconductor suportam mapas de calor complexos e gráficos interativos. Em Python, pandas fornece manipulação de tabelas, enquanto matplotlib, seaborn e plotly cobrem visualização, e scikit-learn oferece um amplo conjunto de métodos multivariados. Ao longo do texto, os autores enfatizam exemplos passo a passo disponibilizados em um GitBook acompanhante, para que leitores possam reproduzir fluxos de trabalho e adaptá-los aos próprios dados. 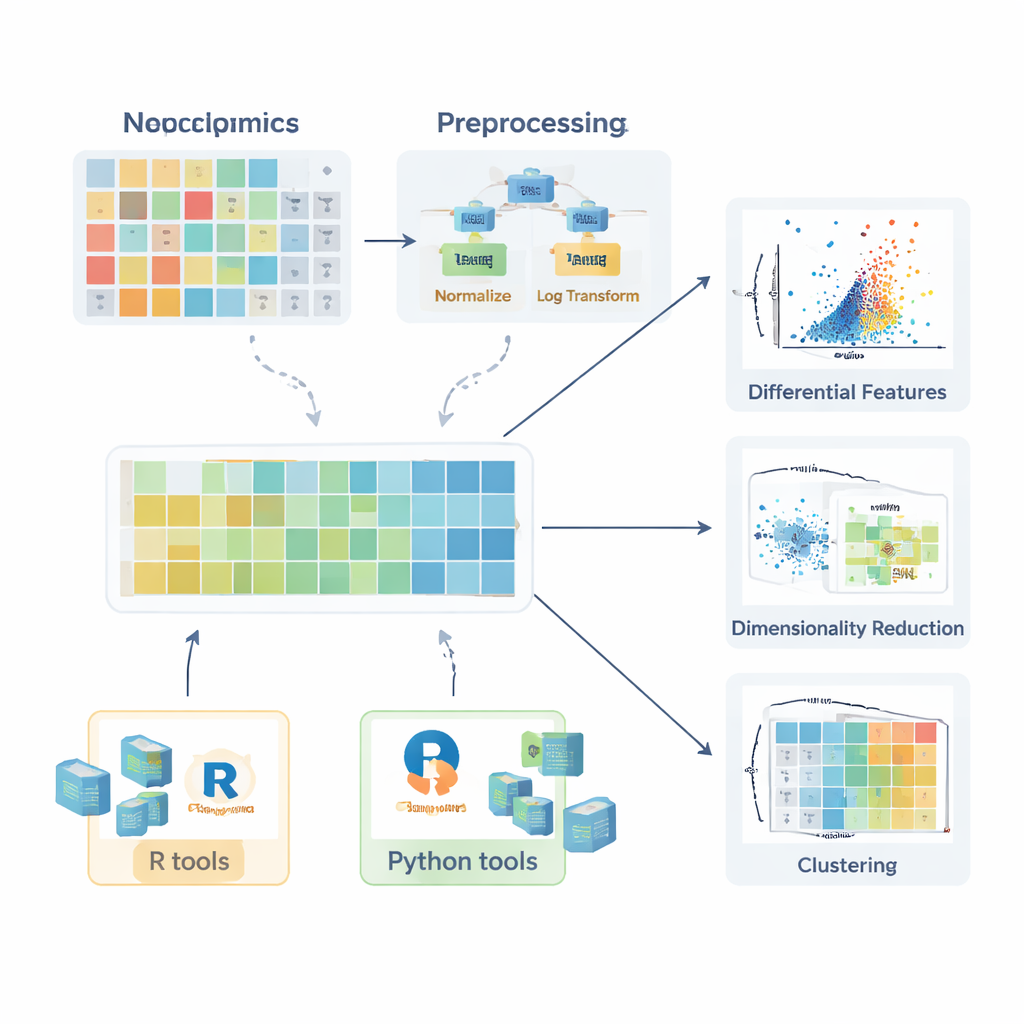
Transformando química complexa em insight confiável
O artigo conclui que a verdadeira promessa da lipidômica e da metabolômica não reside apenas em instrumentos poderosos, mas em como seu resultado é processado e visualizado de forma cuidadosa. Seguindo boas práticas estatísticas, usando ferramentas abertas e bem documentadas em R e Python e apoiando-se em exemplos de código compartilhados, os pesquisadores podem construir fluxos de trabalho robustos e reprodutíveis. Isso aumenta a probabilidade de que padrões encontrados em moléculas minúsculas se traduzam em biomarcadores confiáveis, melhor compreensão dos mecanismos das doenças e abordagens mais personalizadas de medicina que, em última análise, beneficiem os pacientes.
Citação: Idkowiak, J., Dehairs, J., Schwarzerová, J. et al. Best practices and tools in R and Python for statistical processing and visualization of lipidomics and metabolomics data. Nat Commun 16, 8714 (2025). https://doi.org/10.1038/s41467-025-63751-1
Palavras-chave: lipidômica, metabolômica, visualização de dados, programação R, Python