Clear Sky Science · pt
O estado de metilação da fosfatase de proteína 2A impacta a alfa-sinucleinopatia em modelos de camundongos
Por que isso importa para a saúde do cérebro
A doença de Parkinson e distúrbios relacionados roubam lentamente das pessoas a mobilidade, a memória e a independência. Um dos grandes culpados é uma proteína cerebral chamada alfa-sinucleína, que pode se dobrar de forma anômala, aglomerar-se e prejudicar os neurônios. Este estudo faz uma pergunta promissora: em vez de atacar a proteína diretamente, podemos ajustar a própria maquinaria de limpeza do cérebro para evitar que a alfa-sinucleína se torne tóxica?
Uma história sobre uma proteína pegajosa
Na doença de Parkinson e na demência com corpos de Lewy, aglomerados torcidos de alfa-sinucleína se acumulam dentro dos neurônios, formando os clássicos “corpos de Lewy”. Uma marca química específica nessa proteína, adicionada em um sítio chamado serina 129, está fortemente ligada à sua forma mais nociva. Quando essa marca é abundante, a alfa-sinucleína tem maior probabilidade de formar fibrilas rígidas e agregados. O cérebro normalmente equilibra essas marcas usando enzimas que as adicionam e outras que as removem. Como muitas enzimas podem adicionar a marca, bloquear apenas uma delas é pouco provável de funcionar. Em vez disso, os autores focaram na principal família enzimática que remove a marca, chamada fosfatase de proteína 2A, ou PP2A, que atua como uma borracha molecular para essa modificação perigosa.
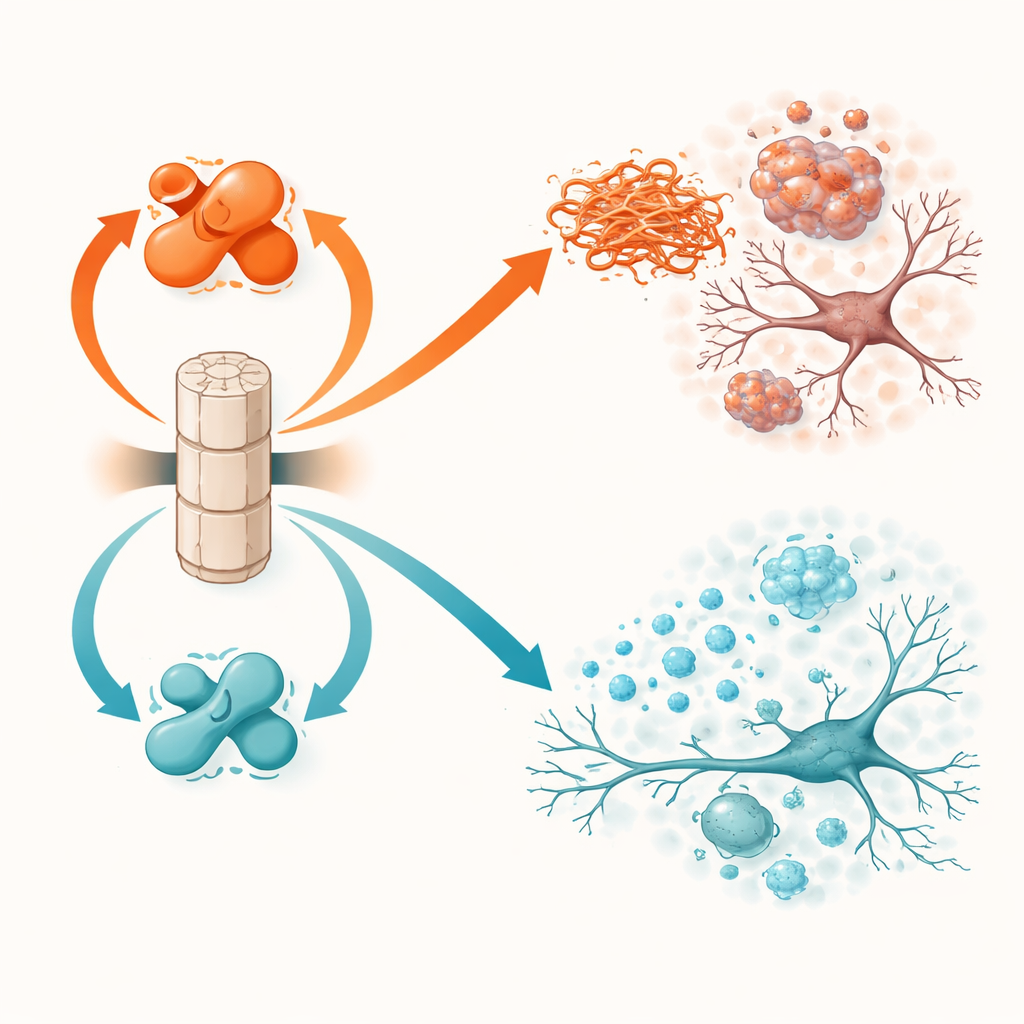
A borracha do cérebro e seus dois interruptores
A PP2A não funciona na potência máxima por padrão. Sua atividade depende de uma pequena marca química, chamada metilação, em uma de suas subunidades. Duas outras proteínas controlam esse interruptor: LCMT-1 adiciona a marca e direciona a PP2A para uma forma mais ativa e protetora, enquanto PME-1 a remove e empurra a PP2A para um estado menos ativo e prejudicial. Trabalhos anteriores em tecido cerebral humano mostraram que, na doença de Parkinson e na demência com corpos de Lewy, LCMT-1 tende a estar reduzida e PME-1 aumentada, deixando a PP2A debilitada. O estudo atual testa diretamente o que acontece quando esses interruptores são manipulados deliberadamente em ambas as direções em camundongos vivos que desenvolvem problemas com alfa-sinucleína.
Testando o equilíbrio em cérebros vivos
Os pesquisadores usaram dois modelos complementares de camundongo. Em um, os animais foram geneticamente modificados para produzir alfa-sinucleína humana por todo o cérebro, desenvolvendo gradualmente aglomerados proteicos e problemas de movimento e memória com a idade. Esses animais foram ainda alterados para superproduzir PME-1 (o interruptor “off” da PP2A) ou LCMT-1 (o interruptor “on” da PP2A) em neurônios do prosencéfalo. No segundo modelo, a equipe injetou fibrilas pré-formadas de alfa-sinucleína no estriado, uma região cerebral profunda envolvida no movimento. Essas fibrilas atuam como sementes, recrutando alfa-sinucleína normal e espalhando a patologia ao longo de meses em camundongos que são, de outra forma, normais ou com alterações enzimáticas. Em ambos os modelos, os cientistas mediram o acúmulo de proteína, a saúde dos neurônios, a inflamação cerebral e o comportamento.
Quando a borracha falha, o dano se espalha
Camundongos que superproduziram PME-1, e portanto tinham PP2A menos ativa, tiveram resultados piores. Em animais transgênicos de alfa-sinucleína, o aumento de PME-1 levou a uma alfa-sinucleína mais fortemente marcada e agregada no córtex e no hipocampo, maior perda da estrutura neuronal, sinais de atividade neuronal mais fracos e ativação mais intensa das células imunes no cérebro. Essas alterações se traduziam em desempenho pior em testes de movimento e em tarefas de aprendizado e memória. No modelo de injeção de fibrilas, a superexpressão de PME-1 permitiu que assembléias tóxicas de alfa-sinucleína se acumulassem e se espalhassem mais extensamente, especialmente para os neurônios produtores de dopamina da substância negra, uma região chave afetada na doença de Parkinson. Esses camundongos exibiram maior perda de fibras dopaminérgicas, inflamação mais intensa e déficits motores e comportamentais mais graves.
Ligando a borracha novamente
A manipulação oposta, superproduzir LCMT-1 para manter a PP2A fortemente metilada e ativa, teve efeitos amplamente protetores. Em camundongos transgênicos de alfa-sinucleína, LCMT-1 reduziu a carga de proteína marcada e agregada a níveis próximos ao normal e preservou tanto a estrutura quanto a atividade dos neurônios. Marcadores inflamatórios estavam mais baixos, e os animais tiveram desempenho mais próximo ao de controles saudáveis em testes de equilíbrio e memória. No modelo de semeação por fibrilas, LCMT-1 limitou tanto o acúmulo local quanto a propagação de longo alcance da alfa-sinucleína tóxica, poupou os neurônios dopaminérgicos da degeneração, reduziu a ativação da microglia e atenuou o declínio na coordenação motora e no comportamento de construção de ninhos. Em todos os experimentos, deslocar a PP2A para seu estado ativo e metilado traduziu de forma consistente benefícios moleculares em proteção funcional.
O que isso pode significar para tratamentos futuros
Para não especialistas, a lição é direta: o cérebro tem uma borracha embutida que pode remover uma marca prejudicial da alfa-sinucleína e impedir que ela se transforme em aglomerados perigosos. Quando essa borracha é enfraquecida, dano, inflamação e sintomas pioram; quando é reforçada, os neurônios ficam protegidos. O estudo fornece evidência direta em animais vivos de que o estado de metilação da PP2A é um controle mestre da toxicidade da alfa-sinucleína e de suas consequências. Isso aponta para uma nova estratégia terapêutica: em vez de perseguir todas as formas nocivas da proteína, fármacos podem ser projetados para inclinar a PP2A e seus reguladores LCMT-1 e PME-1 para um ajuste mais protetor. Essas abordagens exigirão testes de segurança cuidadosos, mas têm potencial para retardar ou prevenir a doença de Parkinson e condições relacionadas ao restaurar a capacidade natural do cérebro de manter a alfa-sinucleína sob controle.
Citação: Maddila, S., Hassanzadeh, K., Liu, J. et al. Protein phosphatase 2A methylation state impacts α-synucleinopathy in mouse models. Cell Death Discov. 12, 172 (2026). https://doi.org/10.1038/s41420-026-03045-7
Palavras-chave: Doença de Parkinson, alfa-sinucleína, fosfatase de proteína 2A, neurodegeneração, inflamação cerebral