Clear Sky Science · pt
Potencializando a atividade de KLF15 em cardiomiócitos: uma abordagem nova para prevenir reprogramação patológica e fibrose via dCas9VPR sem nuclease
Reprogramando o Coração em Falência
A insuficiência cardíaca afeta milhões de pessoas, desenvolvendo-se muitas vezes de forma lenta após anos de hipertensão ou doença valvar. Nessas condições, as células do músculo cardíaco não apenas aumentam de tamanho, mas também ativam um programa genético “fetal” e o coração se enche de tecido cicatricial. Este estudo explora uma nova forma de orientar a maquinaria de controle gênico do próprio coração de volta à saúde — sem cortar o DNA — aumentando de forma moderada um regulador protetor chamado KLF15 nos cardiomiócitos.
Quando as Células Cardíacas Perdem Sua Identidade
No coração adulto saudável, os cardiomiócitos — as células do músculo cardíaco — queimam lipídios de forma eficiente para obter energia e mantêm um padrão estável de atividade gênica. Usando RNA-seq de célula única em camundongos expostos a sobrecarga crônica de pressão, os pesquisadores mapearam como cardiomiócitos individuais mudam à medida que o coração passa da função normal para a dilatação e, finalmente, para a falha. Eles descobriram que um fator de transcrição chamado KLF15, que normalmente mantém o equilíbrio entre metabolismo e crescimento, apresentou a maior alteração de atividade nas células doentes. Conforme o estresse aumentava, os níveis de KLF15 caíam e sua capacidade de conter genes fetais e relacionados ao estresse enfraquecia. Quedas semelhantes de KLF15 foram observadas em corações humanos de pacientes com cardiomiopatia dilatada e hipertrófica, indicando que essa perturbação é conservada entre espécies.
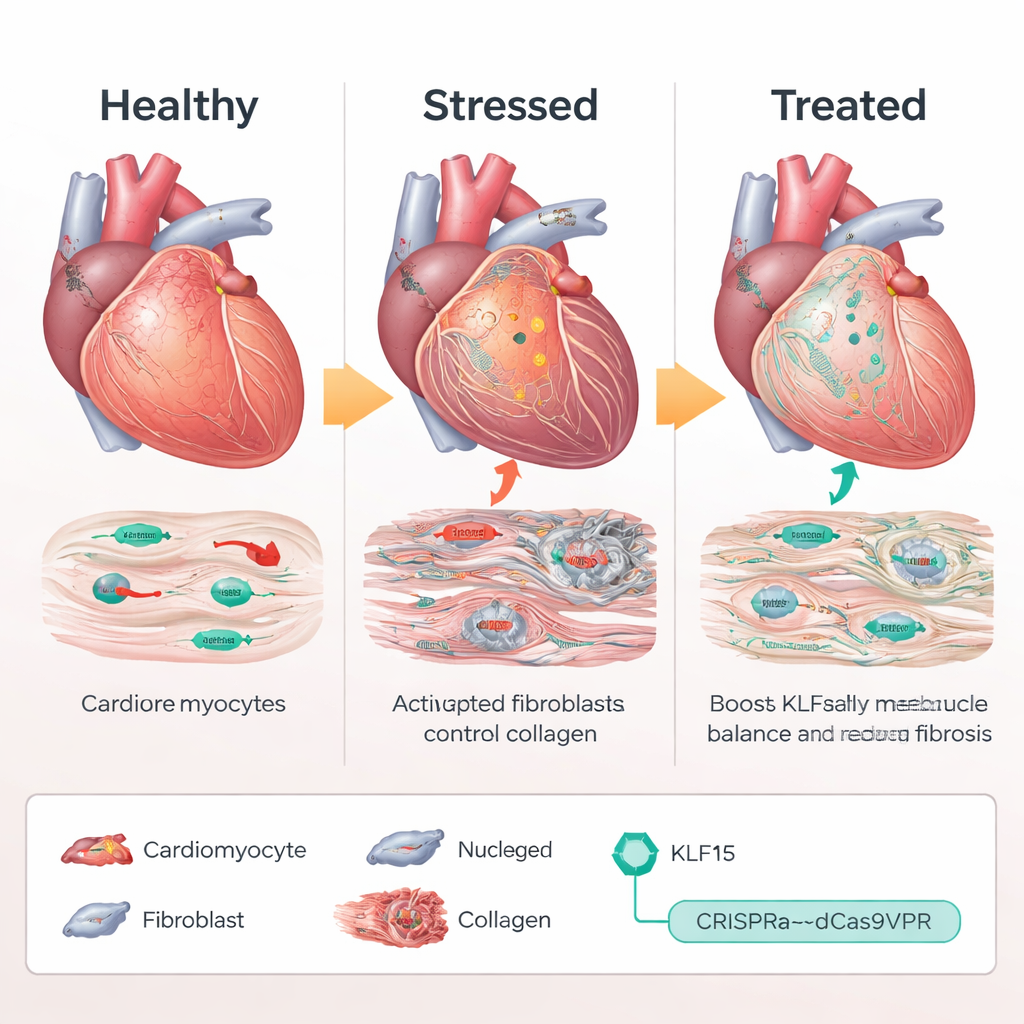
Usando o CRISPR como um Botão de Volume, Não como Tesouras
Em vez de adicionar uma cópia extra do gene KLF15 ou cortar o DNA, a equipe usou um sistema de “ativação” baseado em CRISPR, chamado dCas9VPR, que se liga próximo ao gene Klf15 natural e aumenta sua expressão. Em camundongos geneticamente modificados para expressar esse ativador CRISPR apenas nos cardiomiócitos, os cientistas entregaram RNAs guias por meio de um vírus adeno-associado (AAV9) para direcionar o promotor de Klf15. Sob sobrecarga crônica de pressão, os camundongos que receberam guias ativadores de Klf15 mantiveram níveis próximos do normal de Klf15. Suas células do músculo cardíaco permaneceram menores, a função de bombeamento diminuiu menos e a sobrevivência melhorou em comparação com animais controle. No nível molecular, genes de estresse e fetais foram silenciados, enquanto genes metabólicos e de manejo de cálcio se recuperaram, indicando que o programa transcricional doente foi amplamente redefinido.
Reduzindo a Formação de Cicatriz Através do Diálogo Entre Células
A insuficiência cardíaca é impulsionada não apenas por cardiomiócitos doentes, mas também por fibroblastos, células de suporte que produzem colágeno e formam tecido cicatricial rígido. Análises de célula única e imagens de tecido mostraram que restaurar Klf15 nos cardiomiócitos reduziu a ativação de fibroblastos e a fibrose geral, mesmo que a terapia gênica nunca tenha mirado diretamente os fibroblastos. A equipe rastreou esse efeito até uma proteína secretada chamada AZGP1. Quando Klf15 foi aumentado nos cardiomiócitos, a produção e liberação de AZGP1 aumentaram. Tanto em corações de camundongo quanto em tecidos cardíacos derivados de células-tronco humanas, níveis maiores de AZGP1 atenuaram a via TGF-β / SMAD nos fibroblastos — um motor chave da cicatrização — reduzindo marcadores como α-SMA e POSTN. Importante, a superexpressão de AZGP1 apenas nos cardiomiócitos não reprogramou as células musculares, mostrando que KLF15 protege principalmente os cardiomiócitos de forma direta e usa AZGP1 como mensageiro para conter os fibroblastos.
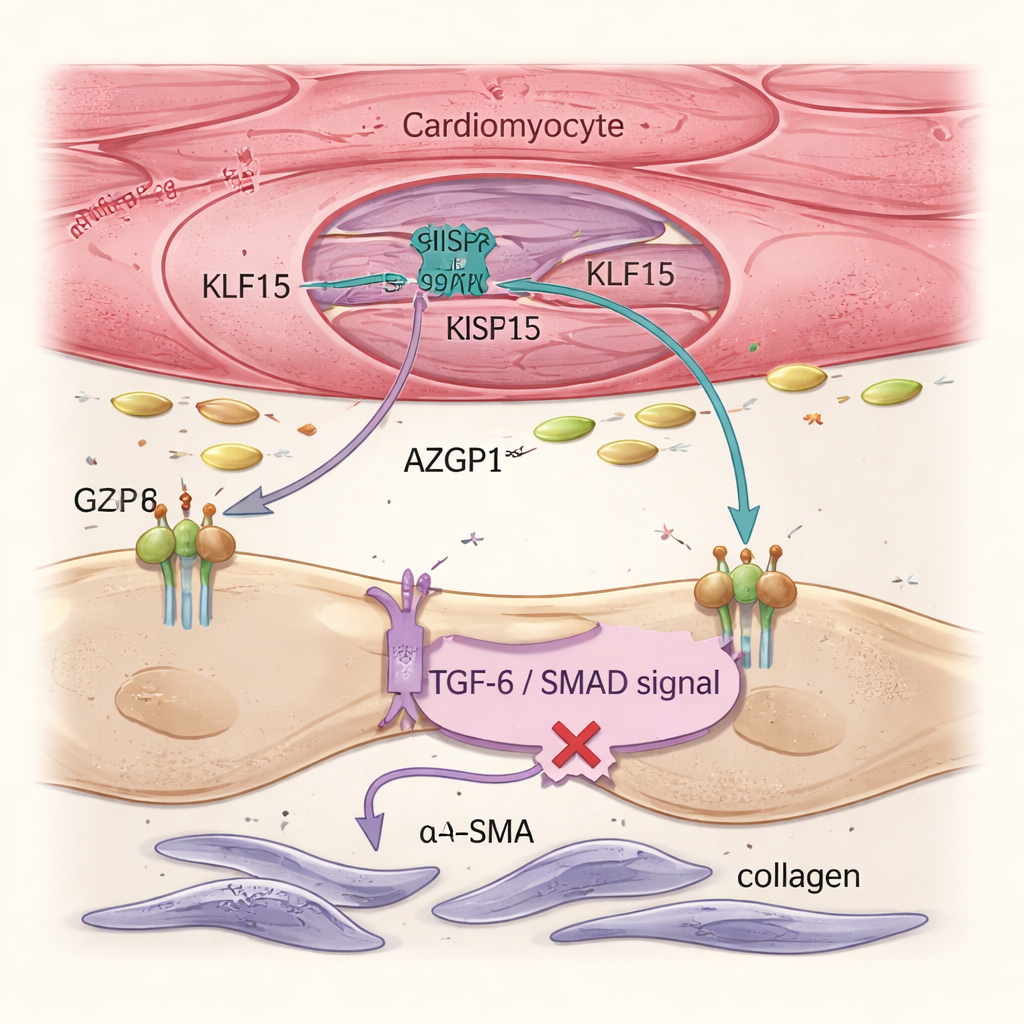
Modelos de Tecidos Humanos Confirmam o Circuito Protetor
Para testar se esses mecanismos se mantêm em células humanas, os pesquisadores usaram cardiomiócitos derivados de células-tronco pluripotentes induzidas crescidos em tecidos cardíacos tridimensionais engenheirados. Quando submetidos a carga mecânica que imita hipertensão, esses tecidos perderam KLF15, ativaram genes de estresse e fetais, tornaram-se mais rígidos e suas contrações enfraqueceram — recapit ulando características da doença. A restauração de KLF15 dirigida por CRISPRa evitou esse declínio, preservou a geração de força e reorganizou a expressão gênica em direção a metabolismo e estrutura maduras. Experimentos detalhados mostraram que TGF-β1, um sinal pró-fibrótico bem conhecido, reduz KLF15 em cardiomiócitos humanos via a via SMAD2/3, ajudando a explicar como o estresse crônico leva ao remodelamento prejudicial. Finalmente, a equipe projetou um sistema CRISPRa compacto, um “mini” baseado em uma variante menor de Cas9 que cabe em um único vetor AAV9 e é dirigido por um promotor específico de cardiomiócitos. Em fatias cardíacas humanas precisas de coração em falha, esse vetor elevou com sucesso os níveis de KLF15 e melhorou o desempenho contratil ao longo de dias em cultura.
Um Roteiro para Terapia Gênica Mais Suave
Para um público não especializado, a mensagem central é que este trabalho mostra como aumentar cuidadosamente um único regulador protetor dentro dos cardiomiócitos pode tanto estabilizar sua identidade quanto enviar sinais que limitam a cicatrização. Ao usar um ativador baseado em CRISPR que não corta o DNA, a abordagem ajusta o próprio gene do coração em vez de inserir um gene artificial. O estudo define uma via TGF-β → KLF15 → AZGP1 que liga o estresse mecânico ao remodelamento prejudicial e demonstra, em camundongos, modelos de células humanas e fatias de tecido cardíaco humano, que restaurar KLF15 pode interromper essa reação em cadeia. Embora ainda esteja em estágio pré-clínico, o sistema CRISPRa compacto e direcionado aos cardiomiócitos apresentado aqui oferece um possível roteiro para tratar formas comuns, não genéticas, de insuficiência cardíaca reprogramando a atividade gênica em vez de reescrever o genoma.
Citação: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
Palavras-chave: insuficiência cardíaca, KLF15, ativação por CRISPR, fibrose cardíaca, AZGP1