Clear Sky Science · pl
Wspomagane przez AI konkurencyjne dokowanie do przesiewów wirtualnych i przewidywania skuteczności związków
Mądrzejsze poszukiwania nowych leków
Poszukiwanie nowych leków przypomina szukanie igły w stogu siana złożonym z milionów cząsteczek. Badanie pokazuje, jak najnowsze osiągnięcia w sztucznej inteligencji mogą przyspieszyć i obniżyć koszt tych poszukiwań, pomagając naukowcom przewidzieć, które cząsteczki najpewniej przyczepią się do białka związanego z chorobą i rzeczywiście zadziałają jako leki. Zamiast testować pojedynczy związek w laboratorium, autorzy używają modeli AI do przeprowadzania wirtualnych pojedynków między cząsteczkami i pozwalają zwycięzcom wypłynąć na wierzch.
Jak AI uczy się rozpoznawać dopasowanie zamka i klucza
Wiele nowoczesnych leków działa, wpasowując się w niewielkie kieszonki na białkach, podobnie jak klucz pasuje do zamka. Tradycyjnie programy komputerowe próbowały przewidzieć to dopasowanie za pomocą równań fizycznych szacujących siły między atomami. W ostatnich latach jednak nowe systemy AI oparte na modelach dyfuzyjnych współskładania — takie jak AlphaFold3 i Boltz — uczyły się na ogromnej liczbie znanych struktur białko–cząsteczka. Systemy te potrafią teraz „wyobrazić sobie”, jak białko i potencjalny lek mogą złożyć się razem w trzech wymiarach, nawet gdy nie istnieje eksperymentalna struktura. Główne pytanie, które stawiają autorzy, brzmi: czy narzędzia AI potrafią zrobić coś więcej niż naszkicować wiarygodne obrazy — czy potrafią też rozróżnić dobre leki od złych?
Prawdziwi wiążący kontra udawacze
Zespół najpierw przetestował 16 dobrze zbadanych białek oraz bardziej złożony enzym bakteryjny, gyrazę DNA. Dla każdego białka poprosili modele AI, by umieściły zarówno znane aktywne inhibitory, jak i zestaw niepowiązanych „off-targetowych” cząsteczek w tym samym miejscu wiążącym. Zamiast polegać na pojedynczej prognozie, obserwowali, jak konsekwentnie AI umieszcza każdą cząsteczkę w wielu powtórzeniach. Prawdziwe inhibitory miały tendencję do powracania do tej samej pozycji i orientacji raz po raz, grupując się w obrębie kilku bilionowych części metra. Cząsteczki nieaktywne dryfowały szerzej i często znajdowały się dalej od kieszonki. Ten prosty pomysł — zbieżność pozycji (pose convergence) — okazał się silnym sygnałem, że związek naprawdę pasuje do celu białkowego.
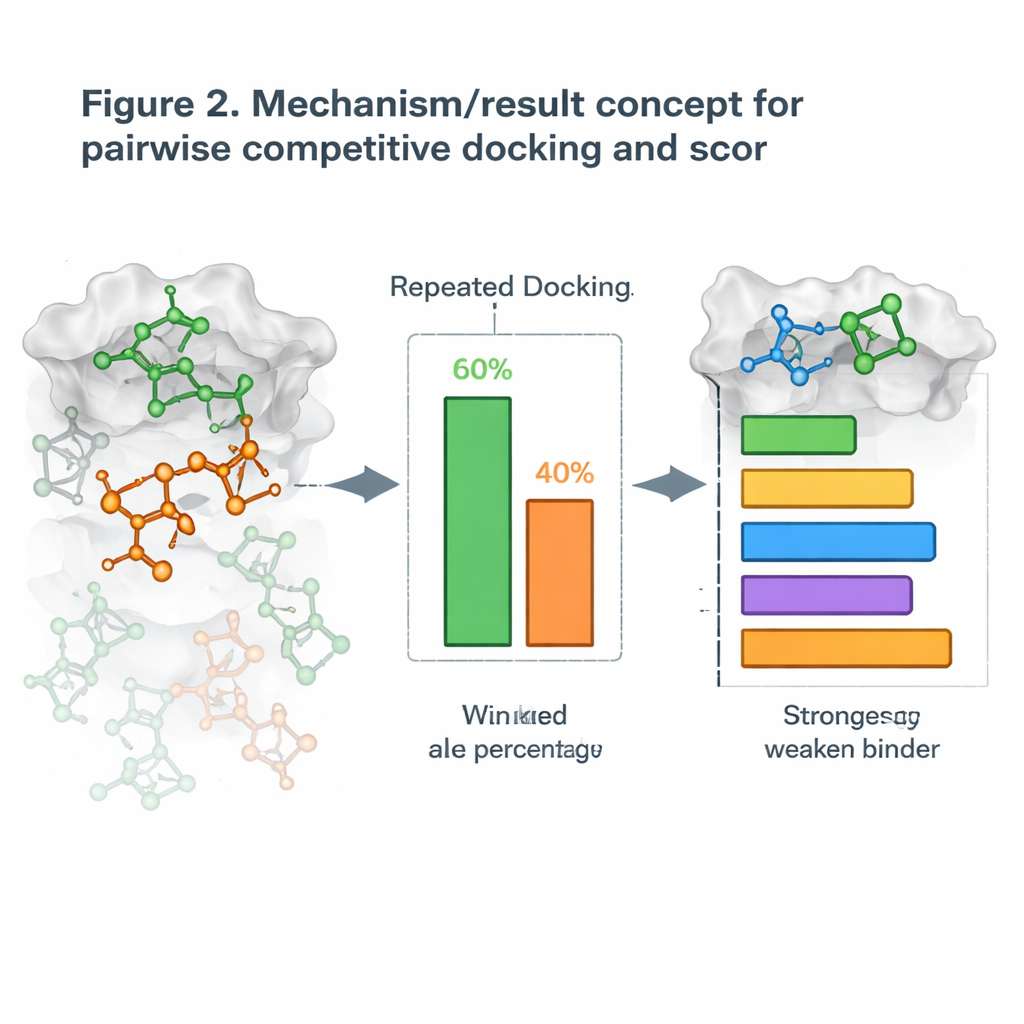
Przekształcanie dokowania w bezpośrednią rywalizację
Na tej podstawie autorzy wprowadzili nową strategię nazwaną parowym konkurencyjnym dokowaniem. Zamiast dokować jedną cząsteczkę na raz, dokują dwie kandydatki jednocześnie z białkiem i pozwalają im „konkurować” o tę samą kieszonkę. Po wielu powtórzeniach cząsteczka, która częściej zajmuje miejsce, zostaje uznana za zwycięzcę tego pojedynku. Uruchamiając wszystkie możliwe parowania, budują tabelę zwycięstw–porazek i obliczają dla każdej cząsteczki Współczynnik Konkurencyjnego Dokowania, podobnie jak ranking zawodników w turnieju każdy z każdym. Gdy te wyniki porównano z rzeczywistymi pomiarami siły blokowania celów przez cząsteczki, rankingi często dobrze się zgadzały, a w niektórych układach białkowych zgodność była niemal doskonała.
Od przesiewów wirtualnych do projektowania lepszych antybiotyków
Gyraza DNA, enzym niezbędny dla bakterii, posłużyła jako szczegółowy przypadek testowy. Białko to ma kilka kieszonek leku celowanych przez różne klasy antybiotyków, w tym szeroko stosowane fluorochinolony. Modele AI zwykle potrafiły umieścić każdą klasę leków w jej poprawnej kieszeni, a konkurencyjne oceny dokowania w przybliżeniu odzwierciedlały ich zmierzone powinowactwa. Autorzy rozszerzyli następnie badanie do przesiewu wirtualnego ponad 3000 zatwierdzonych leków, pytając, które cząsteczki najlepiej rywalizują o miejsce fluorochinolonów. Ich dwuetapowa strategia — najpierw użycie „wszystko naraz” competition, by wybrać prawdopodobnych zwycięzców, a potem filtrowanie według tego, jak ciasno się grupują w kieszeni — znacząco wzbogaciła pulę prawdziwych fluorochinolonów przy odrzuceniu słabszych kandydatów. Na koniec użyli generatora cząsteczek opartego na AI, by zaproponować nowe struktury podobne do fluorochinolonów, i zastosowali konkurencyjne dokowanie, by znaleźć kilka z jeszcze lepiej przewidywanym wiązaniem i akceptowalnymi właściwościami farmakologicznymi.
Obietnice, ograniczenia i co to oznacza dla pacjentów
Badanie pokazuje, że nowoczesne modele AI potrafią zrobić więcej niż narysować wiarygodne struktury białko–lek: uruchomione w ramie konkurencyjnej, mogą pomóc w rankingu związków w sposób często odzwierciedlający rzeczywiste dane eksperymentalne. To nie zastępuje pracy laboratoryjnej — wydajność nadal silnie zależy od konkretnego białka, niektóre kieszonki są źle przewidywane, a modele AI mogą zawodzić w przypadku bardzo dużych lub nietypowych cząsteczek. Jednak w miarę ulepszania tych modeli i ich danych treningowych, podejścia takie jak parowe konkurencyjne dokowanie mogą uczynić wczesne odkrywanie leków znacznie bardziej efektywnym. Dla pacjentów może to w przyszłości przełożyć się na szybsze opracowywanie ukierunkowanych leków, w tym nowych antybiotyków nadążających za opornością bakterii.
Cytowanie: Mirgaux, M., Barcelli, V., Chua, A.C.Y. et al. AI-guided competitive docking for virtual screening and compound efficacy prediction. npj Drug Discov. 3, 6 (2026). https://doi.org/10.1038/s44386-026-00039-4
Słowa kluczowe: AI w odkrywaniu leków, przesiewanie wirtualne, docking molekularny, wiązanie białko-ligand, projektowanie antybiotyków