Clear Sky Science · pl
Energooszczędne obliczenia naukowe z wykorzystaniem rezerwuarów chemicznych
Dlaczego przemiana chemii w obliczenia ma znaczenie
Nowoczesne superkomputery zużywają ogromne ilości energii elektrycznej, symulując klimat, projektując nowe leki czy trenując sztuczną inteligencję. W miarę jak zbliżamy się do fizycznych granic tradycyjnych układów scalonych, uzyskanie większej wydajności na wat staje się trudniejsze i droższe. Artykuł bada radykalnie odmienną drogę: wykorzystanie rzeczywistych reakcji chemicznych jako napędu obliczeń naukowych. Traktując cząsteczki i ich wzajemne oddziaływania jako elementy wykonawcze komputera, autorzy przedstawiają, jak przyszłe maszyny mogłyby rozwiązywać złożone równania przy znacznie niższym zużyciu energii niż współczesny sprzęt cyfrowy.
Od żywych komórek do chemicznych kalkulatorów
Komórki żywe są mistrzami rozwiązywania problemów. Stale żonglują tysiącami reakcji, by się adaptować, rosnąć i przetrwać, a robią to przy zaskakująco niskim zużyciu energii. U podstaw tego działania leżą sieci reakcji chemicznych — powiązane reakcje, których szybkości i stężenia zmieniają się w czasie. Sieci te można opisać równaniami różniczkowymi zwyczajnymi, tym samym językiem matematycznym, którym modeluje się zjawiska od epidemii po przepływy turbulentne. Wniosek stojący za tą pracą jest taki, że skoro chemia już podąża za tymi równaniami, można ją bezpośrednio wykorzystać do przeprowadzania obliczeń, które dziś wykonują krzemowe układy.
Jak równania stają się sieciami reakcji
Autorzy wprowadzają ChemComp, ramy programowe, które przyjmują układ równań różniczkowych i systematycznie przekształcają go w abstrakcyjną sieć reakcji. ChemComp korzysta z nowoczesnych technologii kompilatorowych, aby rozłożyć problem matematyczny na wzorce możliwe do przedstawienia jako zidealizowane reakcje, a następnie organizuje je w sieć z dobrze określonymi gatunkami, połączeniami i stałymi szybkości. Te abstrakcyjne reakcje nie odpowiadają jeszcze rzeczywistym molekułom, ale tworzą plan chemicznego komputera. Ramy mogą potem przeszukiwać bazy danych reakcji biochemicznych w poszukiwaniu rzeczywistych motywów reakcyjnych o podobnym zachowaniu, faworyzując rozwiązania praktyczne, bezpieczne i potencjalnie energooszczędne w warunkach laboratoryjnych.
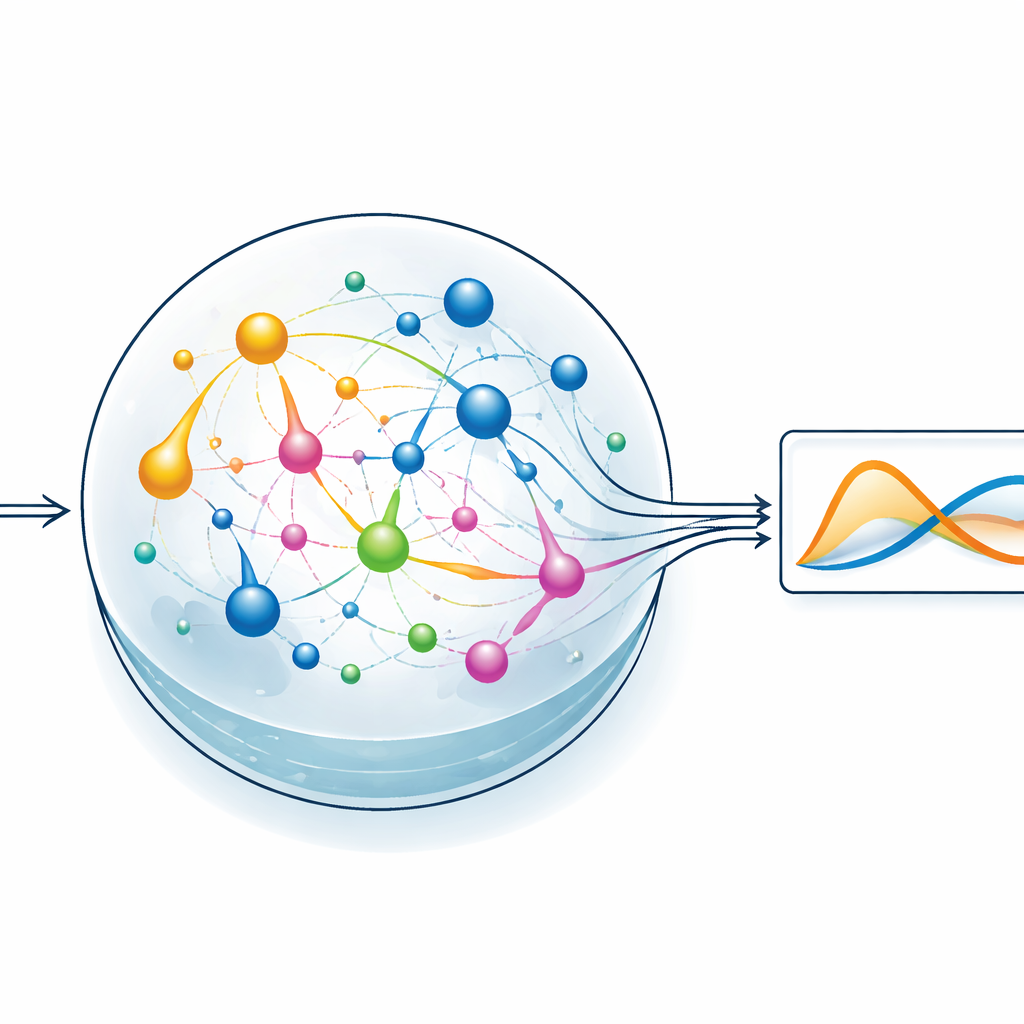
Pozwolić rezerwuarowi chemicznemu wykonać ciężką pracę
Aby przetestować pomysł, zespół skupia się na stylu uczenia maszynowego zwanym reservoir computing. W tym podejściu stały, dynamiczny system przekształca sygnał wejściowy w bogaty, splątany wzorzec aktywności wewnętrznej, a trenowana jest jedynie prosta warstwa odczytu produkująca pożądane wyjście. W wersji ChemComp rezerwuar stanowi zestaw reakcji w dobrze wymieszanym naczyniu; zmieniające się stężenia chemikaliów tworzą stany wewnętrzne. Autorzy kompilują klasyczny układ dwuzmiennych znany jako model Sel’kowa–Schnakenberga — pierwotnie używany do badania oscylacji w metabolizmie — do postaci kandydujących sieci reakcyjnych. Następnie symulują, jak te sieci reagują w czasie, gdy są napędzane przepływem związków chemicznych do i z naczynia, i używają podstawowej regresji liniowej, aby przekształcić przebiegi stężeń w przybliżenie docelowego rozwiązania.
Testowanie prostych i bogatszych sieci chemicznych
Naukowcy porównują dwa kandydujące rezerwuary: jeden z zaledwie dwoma gatunkami chemicznymi i dwiema reakcjami, oraz drugi z pięcioma gatunkami i pięcioma reakcjami. Oba systemy otrzymują odpowiednie koncentracje początkowe i szybkości przepływu, po czym symulowane są ich działanie. Nawet mniejszy układ potrafi w przybliżeniu odtworzyć oscylacyjne zachowanie docelowych równań, ale większa sieć radzi sobie wyraźnie lepiej, zmniejszając błąd podczas treningu i testów. Przeszukując różne początkowe stężenia i stałe szybkości reakcji, autorzy wyznaczają obszary, w których system chemiczny najwierniej odpowiada pożądanej dynamice. Każda reakcja działa efektywnie jak funkcja bazowa w zadaniu dopasowywania krzywej: im więcej zróżnicowanych reakcji jest dostępnych, tym łatwiej przybliżyć złożone zachowanie, kosztem zwiększonej złożoności systemu.
Droga do niskoenergetycznych obliczeń w laboratorium
Ponad symulacjami, artykuł patrzy w stronę praktycznych urządzeń. Omawia, jak wybór reakcji musi równoważyć zużycie energii, sterowalność za pomocą enzymów lub katalizatorów oraz możliwość pomiaru kluczowych gatunków w czasie rzeczywistym, na przykład metodami optycznymi lub elektrochemicznymi. Autorzy sugerują, że przyszłe platformy mikroprzepływowe mogłyby gościć starannie dobrane sieci reakcyjne, z przestrzenną kontrolą wejść i wbudowanym systemem detekcji. Chociaż pozostaje wiele wyzwań inżynieryjnych — od mapowania równań na rzeczywistą chemię po radzenie sobie z szumem i ograniczeniami pomiarowymi — badanie pokazuje, że skromne układy reakcyjne już teraz potrafią emulować rozwiązania sprzężonych równań różniczkowych. Dla czytelnika nieznającego tematu kluczowy przekaz jest taki, że sama chemia może działać jak komputer analogowy, otwierając drogę do obliczeń naukowych opartych na energooszczędnych procesach, które natura doskonaliła przez miliardy lat.
Cytowanie: Johnson, C.G.M., Bohm Agostini, N., Cannon, W.R. et al. Energy-efficient scientific computing using chemical reservoirs. npj Unconv. Comput. 3, 17 (2026). https://doi.org/10.1038/s44335-026-00053-9
Słowa kluczowe: obliczenia chemiczne, energooszczędne obliczenia, reservoir computing, sieci reakcji chemicznych, równania różniczkowe zwyczajne