Clear Sky Science · pl
Kompleksowe ramy do modelowania reaktywności w katalizie heterogenicznej
Dlaczego szybsze projektowanie katalizatorów ma znaczenie
Współczesne społeczeństwo polega na katalizatorach przy produkcji paliw, tworzyw sztucznych, nawozów i niezliczonych produktów codziennego użytku. Tymczasem znalezienie lepszych katalizatorów przypomina często szukanie igły w stogu siana, ponieważ każdy materiał może jednocześnie katalizować tysiące możliwych mikroskopijnych reakcji. W artykule przedstawiono CARE — nowe ramy obliczeniowe, które wykorzystują reguły i uczenie maszynowe do mapowania i symulacji tych splątanych sieci reakcji znacznie szybciej i bardziej kompleksowo niż dotychczas. Dzięki temu mogą one naprowadzać rozwój czystszych technologii energetycznych i bardziej wydajnych procesów chemicznych przy znacznym obniżeniu kosztów obliczeniowych.
Rozplątywanie zatłoczonych ścieżek reakcyjnych
Na powierzchni stałego katalizatora napływające cząsteczki nie podążają po prostu jedną uporządkowaną drogą od substratu do produktu. Zamiast tego przemieszczają się przez labirynt krótkotrwałych pośredników i konkurujących ścieżek. Tradycyjne metody komputerowe polegają na intuicji badacza, by wybrać ograniczony zestaw możliwych kroków, a następnie ocenić ich energie za pomocą obliczeń kwantowych. To działa dla małych sieci, ale szybko zawodzi w miarę wzrostu złożoności, pomijając rzadkie ścieżki, które mogą rządzić długoterminową aktywnością, dezaktywacją lub selektywnością. CARE podchodzi do tego wyzwania, automatycznie konstrukując bardzo duże sieci reakcji z prostych reguł budulcowych, zapewniając włączenie wszystkich prawdopodobnych zdarzeń łamania i tworzenia wiązań między węglem, wodorem i tlenem, nawet tych, które chemicy zwykle by odrzucili.
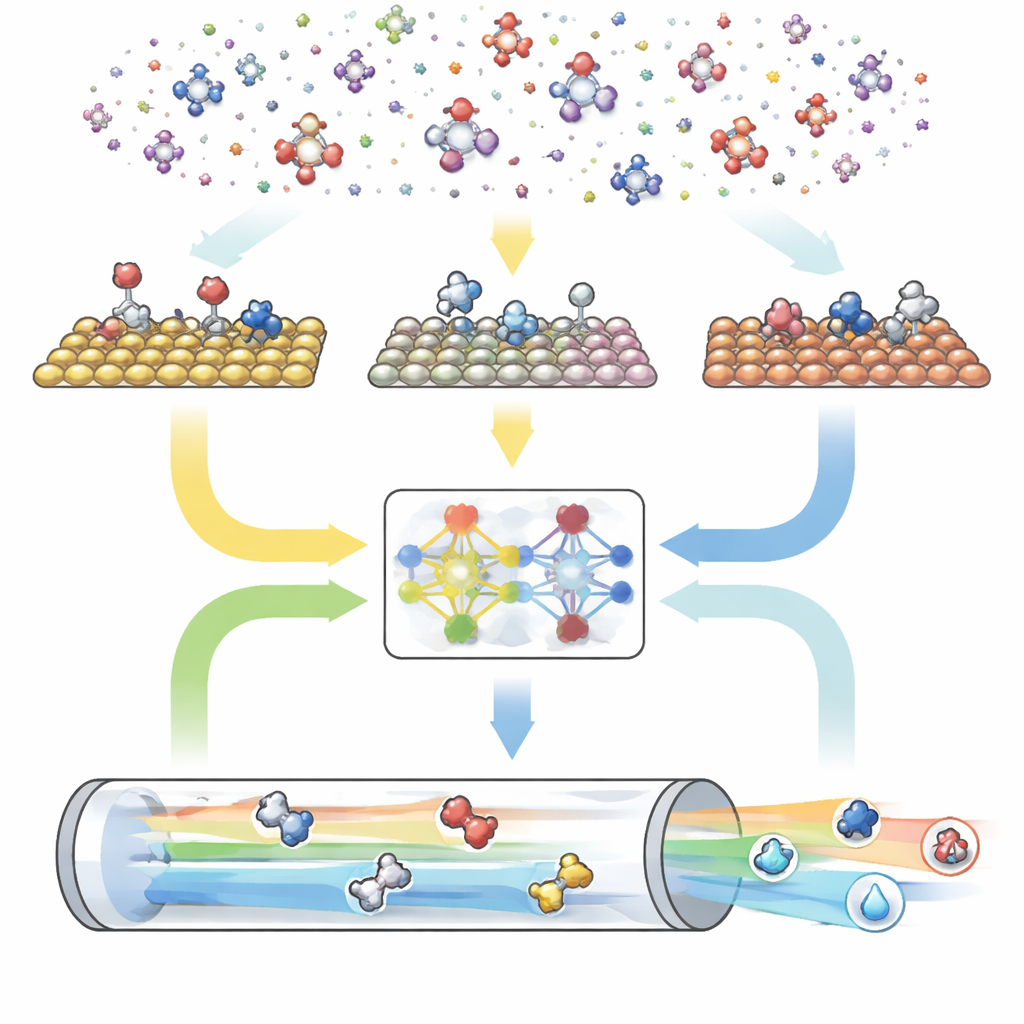
Trójdzielny cyfrowy silnik reakcji
CARE jest zbudowane jako end-to-end pipeline z trzema głównymi modułami. Po pierwsze, generator oparty na regułach definiuje „przestrzeń chemiczną” przez wybór maksymalnej liczby atomów węgla i tlenu, a następnie stosuje proste szablony do wygenerowania wszystkich pasujących cząsteczek i ich form związanych z powierzchnią. Po drugie, moduł oceny energii korzysta z nowoczesnych modeli uczenia maszynowego — w szczególności z grafowej sieci neuronowej nazwanej GAME-Net-UQ — aby oszacować energie pośredników i stanów przejściowych na różnych powierzchniach metalicznych. Model traktuje każdą strukturę jako sieć atomów i wiązań, zwraca zarówno energię, jak i niepewność, i jest dokładny do kilku dziesiątych elektronowolta przy zachowaniu lekkości i szybkości. Po trzecie, solver mikrokinetyczny wykorzystuje te energie do obliczenia, jak wszystkie reakcje przebiegają razem w realistycznych warunkach temperatury, ciśnienia, napięcia i pH, przewidując ogólne szybkości reakcji, pokrycie powierzchni i selektywność produktów.
Testy w praktyce: cząsteczki paliwowe i chemia klimatu
Aby pokazać, że CARE nie jest jedynie ćwiczeniem teoretycznym, autorzy zastosowali je do trzech przemysłowo istotnych problemów o rosnącej trudności. Dla dekompozycji metanolu — reakcji ważnej dla magazynowania wodoru — wygenerowali umiarkowaną sieć i ocenili ją dla wielu katalizatorów metalicznych i ścianek krystalicznych. CARE odtwarza znajomy trend „wulkaniczny” aktywności i poprawnie identyfikuje ruten jako jednego z najlepszych wykonawców, zgodnie z eksperymentami, lecz przy ułamku czasu obliczeniowego potrzebnego dla pełnych obliczeń kwantowych. Następnie przeszli do elektrochemicznej konwersji dwutlenku węgla na miedzi, koncentrując się na powstawaniu produktów trójwęglowych, takich jak 1-propanol i propylen. Dzięki uwzględnieniu specjalnych kroków biorących pod uwagę protony, elektrony i warunki roztworu, CARE uchwyciło, jak pH i przyłożone napięcie przesuwają ścieżki i poprawnie przewidziało, że 1-propanol jest faworyzowany względem propylenku, zgodnie ze szczegółowymi wcześniejszymi badaniami.
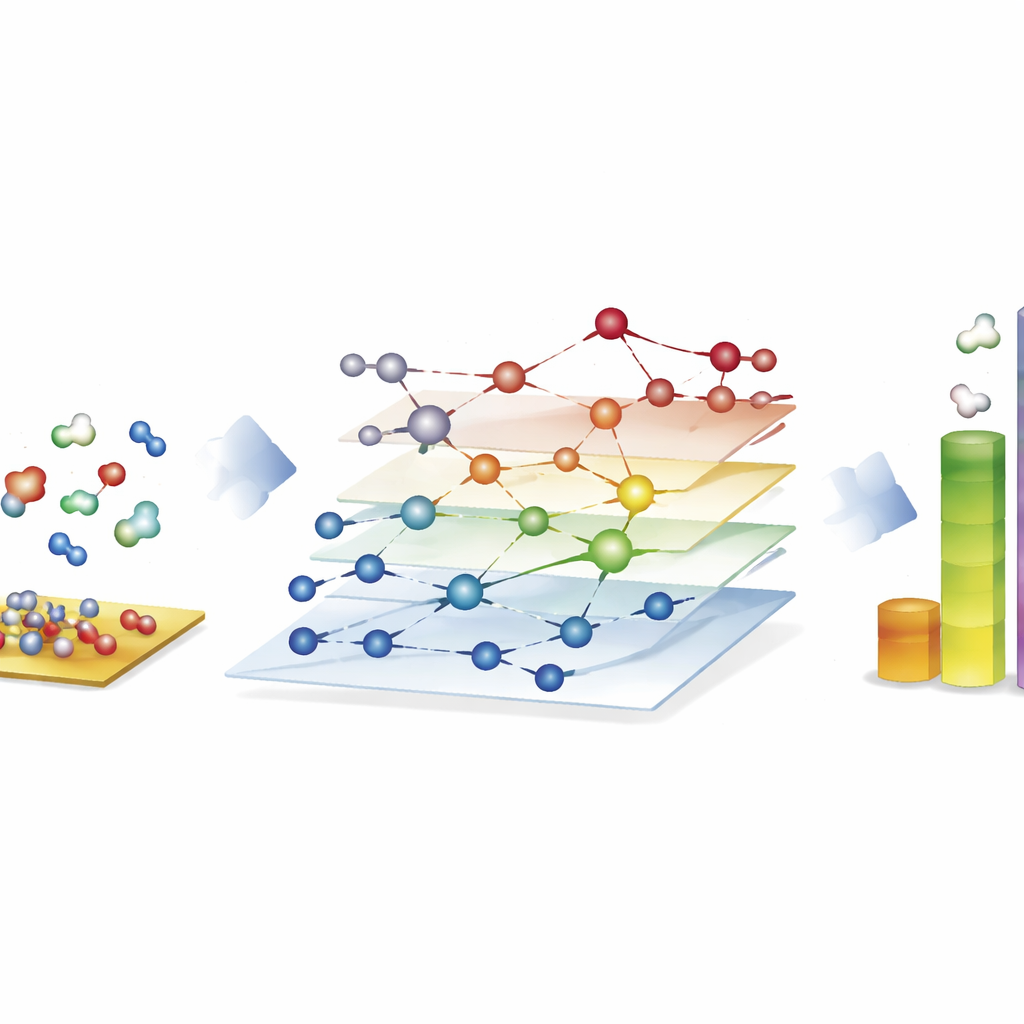
Badanie ogromnych sieci reakcyjnych dla paliw syntetycznych
Najbardziej uderzającą demonstracją jest proces Fischera–Tropscha, który przekształca mieszaniny tlenku węgla i wodoru w długie łańcuchy węglowodorów na potrzeby paliw i chemikaliów. Tutaj autorzy skonstruowali sieci z prawie 40 000 gatunkami powierzchniowymi i około 370 000 reakcji elementarnych — daleko poza zasięgiem, które tradycyjne badania oparte na obliczeniach kwantowych mogłyby w pełni zbadać. Korzystając z CARE, ocenili wszystkie pośredniki i kluczowe bariery reakcyjne na powierzchniach kobaltu, żelaza, niklu i rutenu w ciągu zaledwie kilku godzin na standardowym sprzęcie, co stanowi przyspieszenie rzędu miliona razy w porównaniu z bezpośrednimi obliczeniami kwantowymi. Symulacje mikrokinetyczne tych sieci odtwarzają znane trendy: kobalt i żelazo preferencyjnie tworzą dłuższe łańcuchy węglowodorowe, żelazo wytwarza więcej dwutlenku węgla przez reakcje uboczne, a nikiel skłania się ku silniejszemu uwodorowaniu. Chociaż niektóre szczegóły, takie jak wydajność metanu, pozostają niedoskonałe, ramy ujawniają, które kroki tworzenia wiązań dominują przy wzroście łańcucha i wskazują, gdzie modele nadal wymagają dopracowania.
Co to oznacza dla przyszłych katalizatorów
Dla osób niebędących specjalistami kluczowy przekaz jest taki, że CARE daje praktyczny sposób eksploracji ogromnych przestrzeni reakcyjnych na powierzchniach katalitycznych, które wcześniej były poza zasięgiem. Automatyzując generowanie sieci, podłączając szybkie modele „zastępcze” uczenia maszynowego do obliczeń kwantowych i efektywnie rozwiązując powstałą kinetykę, system potrafi klasyfikować kandydatów na katalizatory, identyfikować obiecujące warunki pracy i odkrywać nieoczekiwane ścieżki przy znacznie mniejszym uprzedzeniu ludzkim i koszcie obliczeniowym. Autorzy zauważają istniejące wyzwania — takie jak lepsze radzenie sobie z zatłoczonymi powierzchniami, wpływem rozpuszczalnika czy jeszcze większymi sieciami — jednak praca wskazuje drogę ku przyszłości, w której komputery będą mogły szybko przesiewać złożone reakcje, od redukcji dwutlenku węgla po recykling plastiku i ulepszanie biomasy, kierując eksperymenty ku najbardziej obiecującym pomysłom zamiast polegać na metodzie prób i błędów.
Cytowanie: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
Słowa kluczowe: kataliza heterogeniczna, sieci reakcji, uczenie maszynowe, mikrokinetyczne modelowanie, synteza Fischera–Tropscha