Clear Sky Science · pl
Integracyjna ocena genomowa i literaturowa kardiomiopatii arytmogennej związanej z desmogleiną 2 z walidacją na kohorcie włoskiej
Dlaczego ten gen serca ma znaczenie dla rodzin
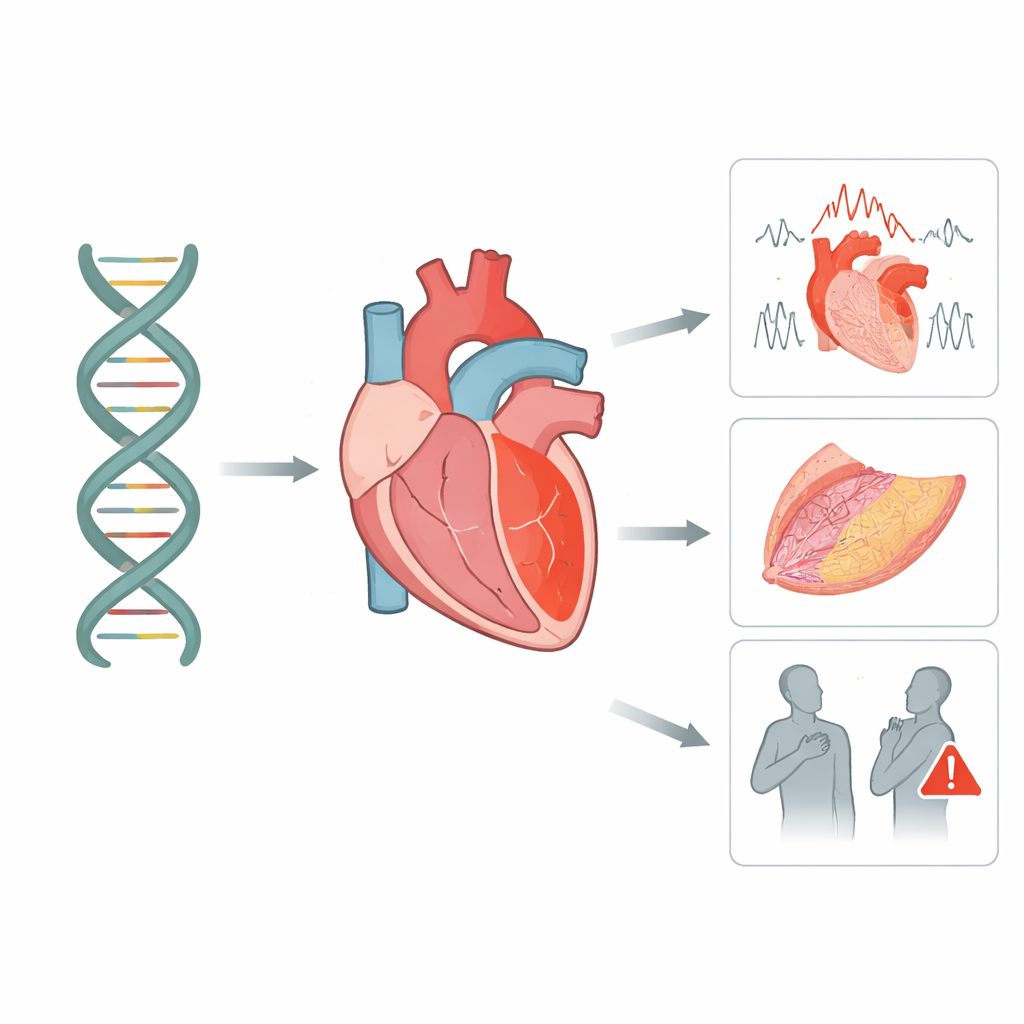
Wiele nagłych problemów sercowych u młodych i pozornie zdrowych osób nie jest przypadkowych — są zapisane, przynajmniej w części, w ich DNA. Ten artykuł bada kluczowe białko „spajające” serce, nazywane desmogleiną‑2, i pokazuje, jak niewielkie zmiany w jego genie mogą osłabić mięsień sercowy, zaburzyć rytm elektryczny serca i zwiększyć ryzyko niebezpiecznych zdarzeń. Łącząc duże bazy danych genetycznych z dokładnie obserwowaną włoską grupą pacjentów, badacze dają rodzinom jaśniejsze odpowiedzi na pytanie, co naprawdę oznacza wynik testu tego genu.
Mechaniczna „spinka” serca
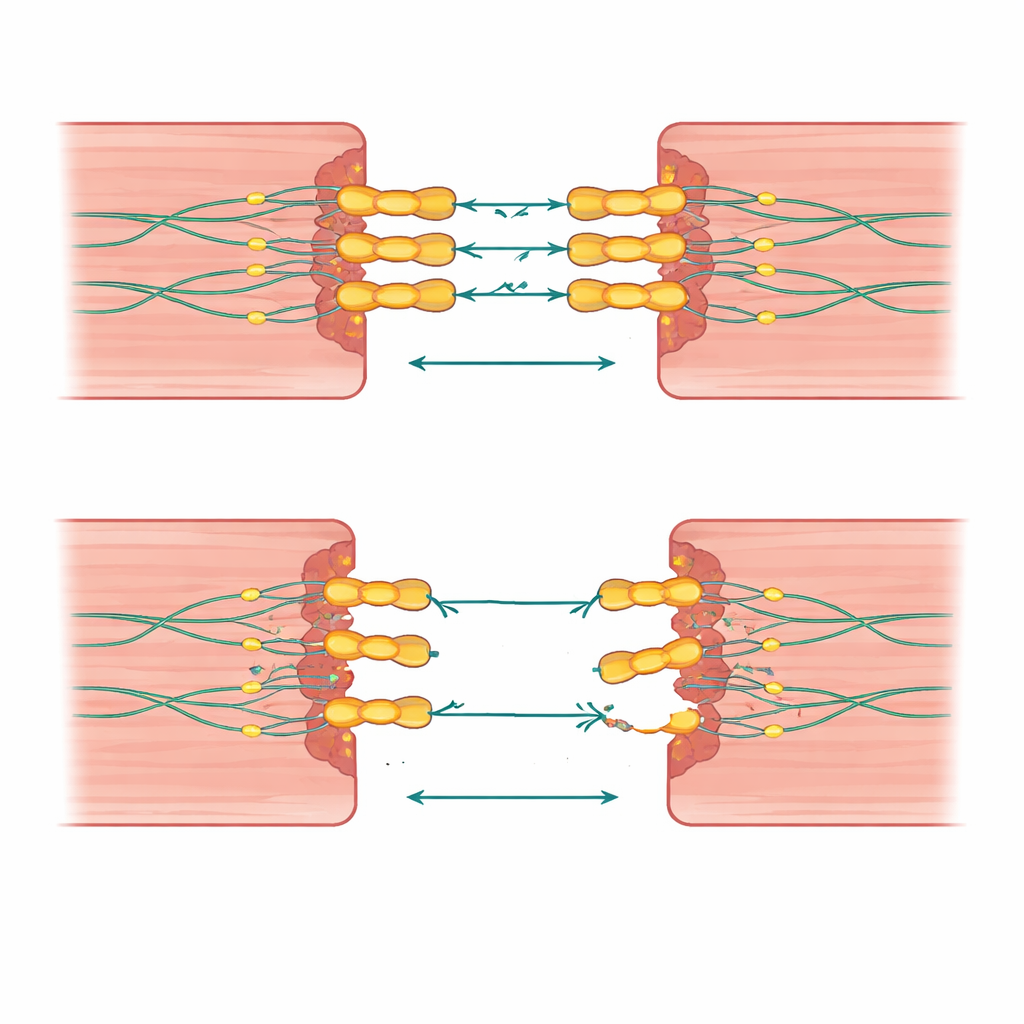
Komórki mięśnia sercowego muszą mocno przylegać do siebie, bijąc miliony razy w ciągu życia. Desmogleina‑2 jest częścią mikroskopowej struktury podobnej do nitów, która łączy sąsiednie komórki, aby mogły ciągnąć razem. Autorzy wyjaśniają, jak to białko rozciąga się od zewnątrz komórki, gdzie chwyta odpowiadającego partnera na sąsiedniej komórce, do wnętrza, gdzie zaczepia się o podporowy szkielet. Ponieważ desmogleina‑2 jest jedynym przedstawicielem swojej rodziny obecnym w komórkach serca, poważne uszkodzenie jej funkcji nie może być skompensowane przez substytut, co czyni serce szczególnie podatnym.
Rozróżnianie istotnych zmian genowych od szumu
Nowoczesne sekwencjonowanie wykrywa tysiące różnic w genie desmogleiny‑2 w populacji, ale większość z nich nie powoduje choroby. Zespół systematycznie przejrzał 115 opublikowanych badań i wykorzystał dwie duże publiczne bazy danych, które łącznie wymieniały ponad 5 000 wariantów. Stosując szeroko akceptowane reguły genetyki medycznej, ponownie sklasyfikowali każdą zmianę pod kątem prawdopodobieństwa jej szkodliwości. Stwierdzili, że naprawdę uszkadzające warianty grupują się w określonych rejonach białka — szczególnie w zewnętrznych segmentach wymagających wapnia do utworzenia sztywnego mostka między komórkami, w krótkim odcinku, który musi zostać przecięty, aby białko dojrzało, oraz w wewnętrznym regionie, który zaczepia się o inne kluczowe białko serca. Wiele innych zmian pozostało „niepewnych”, ale pewna ich część wykazała silne wskazówki znaczenia i została oznaczona do dokładniejszego monitorowania.
Co ujawnia włoska grupa pacjentów
Aby zobaczyć, jak te wzorce genetyczne przekładają się na rzeczywistych ludzi, badacze przeanalizowali 95 osób we Włoszech, które nosiły warianty desmogleiny‑2 i zostały poddane szczegółowej ocenie obejmującej obrazowanie, badania rytmu serca i długoterminową obserwację. Około połowa spełniała surowe kryteria kardiomiopatii arytmogennej, stanu, w którym części mięśnia sercowego stopniowo zastępowane są blizną i tkanką tłuszczową, co sprzyja niebezpiecznym zaburzeniom rytmu. Wśród krewnych będących nosicielami wariantu tylko około cztery na dziesięć faktycznie wykazywało oznaki choroby, co podkreśla, że pozytywny wynik testu genetycznego nie gwarantuje choroby, lecz sygnalizuje konieczność uważnego monitorowania. Osoby z wyraźną chorobą miały istotne obciążenie poważnymi zdarzeniami rytmu, podczas gdy przeszczepy i zgony były rzadsze, lecz nadal występowały.
Kiedy pojedynczy cios nie wystarcza
Uderzająca obserwacja z tej pracy jest taka, że liczba i kombinacja zmian genowych mają znaczenie. Osoby, które odziedziczyły dwie wadliwe kopie desmogleiny‑2, albo jedną zmianę w desmogleinie‑2 plus zmianę w powiązanym genie „spajającym” serce, miały tendencję do zachorowań w młodszym wieku i wykazywały bardziej rozległe uszkodzenia obu stron serca. Niektóre rodziny miały duże delecje lub duplikacje usuwające albo podwajające nie tylko desmogleinę‑2, ale także sąsiednie geny, co ponownie wiązało te zmiany z agresywną postacią choroby i skupiskami nagłych zgonów. W przeciwieństwie do tego, wielu krewnych z pojedynczą zmianą miało łagodne lub żadne objawy, co sugeruje, że tło genetyczne i czynniki życiowe, takie jak aktywność fizyczna, mogą przesuwać równowagę między cichym ryzykiem a ujawnioną chorobą.
Od kształtu białka do ryzyka pacjenta
Aby połączyć kod DNA z efektem fizycznym, zespół wykorzystał zaawansowane modele 3D białka, by zobaczyć, jak konkretne substytucje mogą poluzować rusztowanie desmogleiny‑2. Zmiany, które zniekształcały pętle wiążące wapń lub łamały kluczowe punkty zaczepienia, przewidywano jako destabilizujące białko i osłabiające przyleganie komórka‑do‑komórki. Te strukturalne wskazówki zostały włączone do systemu klasyfikacji, co pomogło przesunąć niektóre graniczne warianty w stronę uznania ich za bardziej prawdopodobnie szkodliwe lub bardziej prawdopodobnie nieszkodliwe. To pomostowanie między modelowaniem molekularnym a danymi klinicznymi przesuwa badania genetyczne poza proste czytanie kodu w kierunku bardziej funkcjonalnego rozumienia.
Co to oznacza dla pacjentów i rodzin
Dla rodzin dotkniętych kardiomiopatią arytmogenną to badanie przynosi zarówno ostrożność, jak i wskazówki. Pokazuje, że nie każdy wariant desmogleiny‑2 jest wyrokiem ciężkiej choroby serca, ale że pewne wzorce — zwłaszcza wielokrotne trafienia lub zmiany w krytycznych regionach białka — wiążą się z wcześniejszymi i poważniejszymi problemami. Autorzy argumentują, że osoby noszące te warianty nie powinny być lekceważone jako „zdrowe dopóki nie udowodniono inaczej”, lecz należy je obserwować przez całe życie z dostosowanymi kontrolami rytmu i badaniami obrazowymi. Ich integracyjne podejście — łączące genetykę big‑data, szczegółowe badania rodzinne i strukturę białka — wskazuje drogę do bardziej precyzyjnych oszacowań ryzyka i pewniejszego doradztwa, gdy zmiana w desmogleinie‑2 pojawi się w teście genetycznym.
Cytowanie: Pinci, S., Celeghin, R., Martini, M. et al. Integrative genomic and literature assessment of desmoglein 2-related arrhythmogenic cardiomyopathy with Italian cohort validation. Commun Med 6, 145 (2026). https://doi.org/10.1038/s43856-026-01416-w
Słowa kluczowe: kardiomiopatia arytmogenna, desmogleina-2, dziedziczne choroby serca, ryzyko genetyczne, nagła śmierć sercowa