Clear Sky Science · pl
Dyfuzja warunkowa z lokalnościowo‑świadomym dopasowaniem modalnym do generowania zróżnicowanych zespołów konformacyjnych białek
Dlaczego ruch białek ma znaczenie
Białka w naszych komórkach nie są sztywnymi rzeźbami; zachowują się bardziej jak małe, elastyczne maszyny, które nieustannie zmieniają swoje kształty. Te zmiany kształtu mogą kontrolować, jak enzymy przyspieszają reakcje, jak receptory reagują na leki i jak sygnały przepływają w komórce. Jednak większość znanych obrazów białek pokazuje tylko jedno „migawkowe” ujęcie, pomijając bogaty zespół form, które rzeczywiście istnieją. W artykule wprowadzono Mac‑Diff — metodę sztucznej inteligencji, która potrafi szybko wygenerować wiele realistycznych kształtów dla danego białka, pomagając naukowcom zobaczyć nie tylko jak białko wygląda, ale jak „oddycha” i się porusza.
Od pojedynczych migawek do poruszających się zespołów
Przez dekady badacze polegali na mozolnych eksperymentach lub długotrwałych symulacjach dynamiki molekularnej, aby badać ruch białek — obie drogi bywają czasochłonne i kosztowne. Przełomowe narzędzia takie jak AlphaFold2 przewidują teraz najprawdopodobniejszą strukturę 3D białka bezpośrednio z sekwencji aminokwasowej, ale zwykle zwracają tylko jedną lub kilka preferowanych form. Wiele białek, zwłaszcza zaangażowanych w przekazywanie sygnałów i regulację allosteryczną, naturalnie zajmuje wiele luźno zdefiniowanych stanów. Autorzy twierdzą, że aby naprawdę zrozumieć, jak takie białka działają — i projektować leki, które wiążą mniej oczywiste, przejściowe formy — potrzebujemy sposobu generowania całych zespołów prawdopodobnych konformacji, a nie tylko jednej najlepszej zgadywanki.
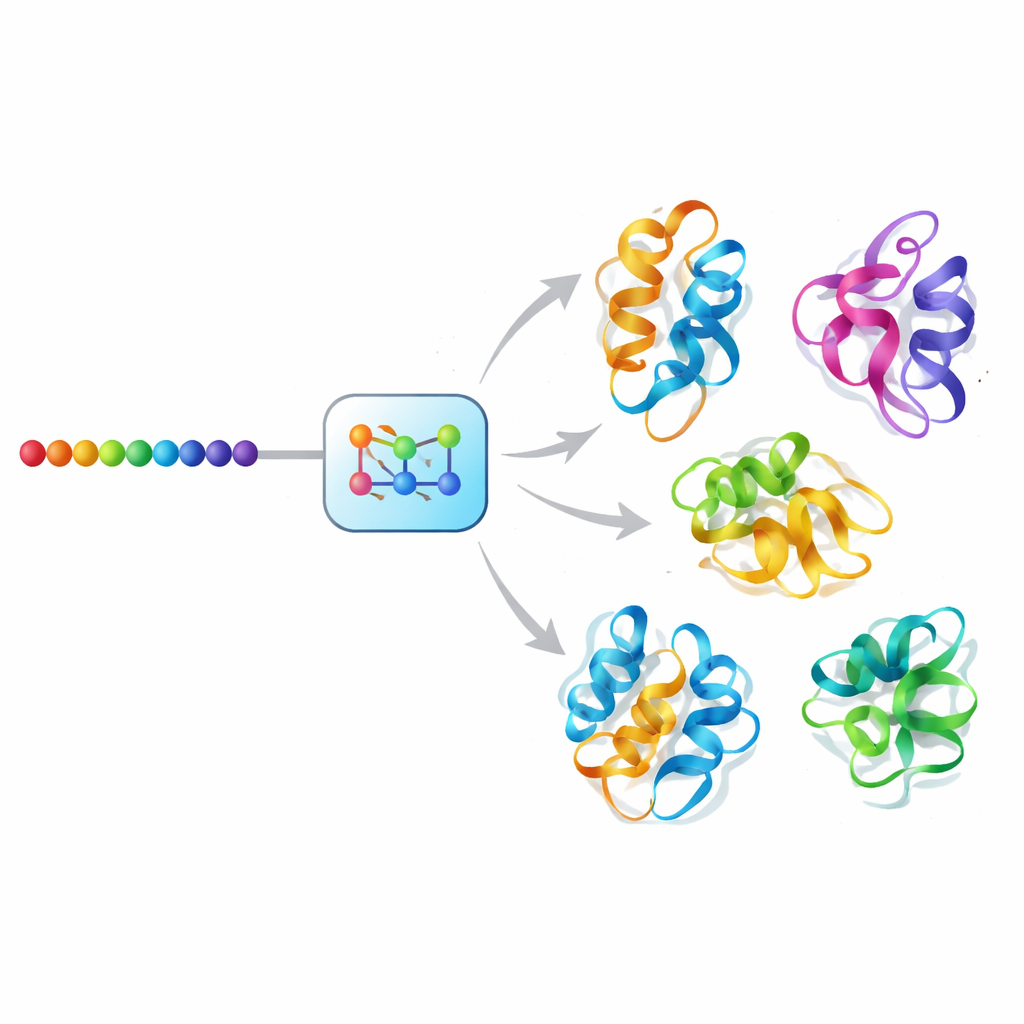
AI‑podejście „dyfuzyjne” do ruchu białek
Mac‑Diff stawia czoła temu wyzwaniu używając generatywnego modelu w stylu dyfuzji, klasy AI, która napędzała niedawne postępy w syntezie obrazów. Zamiast odszumiania fotografii, Mac‑Diff odszumia abstrakcyjne, geometryczne opisy kręgosłupów białkowych. Model reprezentuje białko jako siatkę relacji parowych między resztami — odległości i kąty niewrażliwe na obrót i translację całej cząsteczki. W kroku „do przodu” system stopniowo dodaje szum do tych wzorców geometrycznych, aż przypominają losowy szum. W kroku odwrotnym uczy się usuwać szum krok po kroku, kierowany sekwencją aminokwasową białka, aż znowu pojawią się spójne geometrie zgodne z 3D, które następnie można przekonwertować na pełne modele atomowe za pomocą standardowego oprogramowania do budowy struktur.
Pozwalając sekwencji rozmawiać z strukturą lokalnie
Kluczowa innowacja polega na sposobie łączenia liniowej sekwencji reszt z ich lokalnymi 3D‑sąsiadami. Proste pozwolenie, by każda reszta zwracała uwagę na każdą inną resztę, jak w modelach tekst→obraz, rozmyłoby istotne ograniczenia fizyczne. Zamiast tego autorzy wprowadzają mechanizm uwagi „świadomy lokalności”, który koncentruje się na niewielkim, prawdopodobnym sąsiedztwie partnerów interakcji dla każdej reszty. Aby oszacować te sąsiedztwa, Mac‑Diff wykorzystuje trzy składniki: model języka białkowego ESM‑2, który koduje biochemiczny kontekst każdej reszty; mapę kontaktów sugerującą, które pary reszt mogą być blisko siebie; oraz prostą regułę faworyzującą reszty bliskie sobie wzdłuż łańcucha. Sygnały te są łączone tak, by podczas odszumiania model preferencyjnie korzystał z informacji od reszt będących fizycznie prawdopodobnymi partnerami, co wyostrza jego zdolność do odbudowy realistycznych, elastycznych struktur.
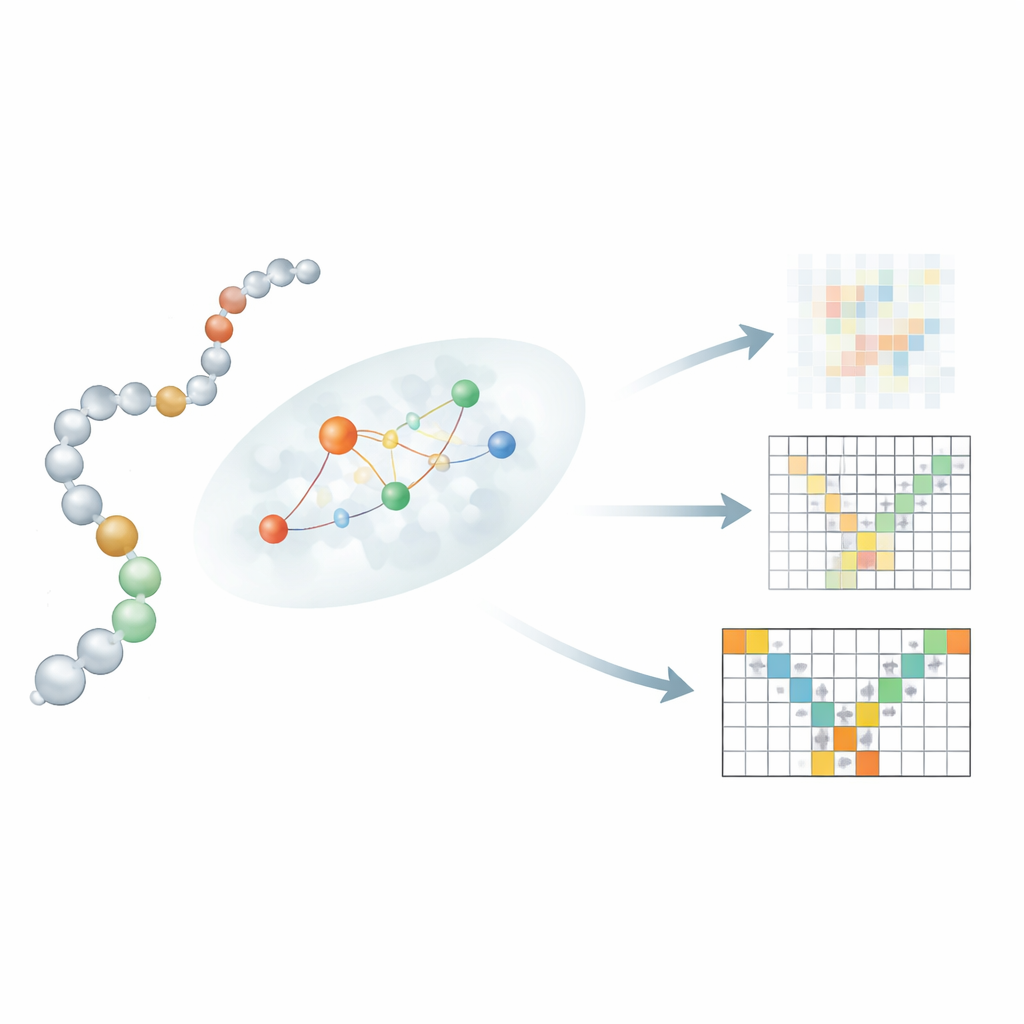
Testy wobec długich symulacji i białek zmieniających kształt
Naukowcy przetestowali Mac‑Diff na dwóch wymagających frontach. Po pierwsze sprawdzili, czy potrafi odtworzyć szerokie rozkłady kształtów obserwowane w długich, starannie obliczonych symulacjach dynamiki molekularnej szybkich fałdujących się białek oraz klasycznego białka referencyjnego BPTI. W kilku miarach porównujących właściwości statystyczne generowanych zespołów z danymi symulacyjnymi — takich jak rozkłady odległości wewnątrz białka i ogólna zwartość — Mac‑Diff dorównał lub przewyższył konkurencyjne metody AI, jednocześnie generując szerszą różnorodność konformacji. Ujął większość kluczowych stanów „metastabilnych” zidentyfikowanych w symulacjach i odtworzył wzorce elastyczności na poziomie reszt z wysoką korelacją, co wskazuje, że jego zespoły odzwierciedlają zarówno globalne fałdowania, jak i lokalne wibracje w realistyczny sposób.
Odkrywanie ukrytych stanów funkcjonalnych
Po drugie zespół wystawił Mac‑Diff na próbę z białkami, które znane są z przyjmowania bardzo różnych kształtów podczas wykonywania swoich funkcji, w tym enzymu adenylanowej kinazy, który przełącza się między formami otwartą i zamkniętą podczas metabolizmu energetycznego, oraz przygotowanego zestawu 40 białek z dwiema eksperymentalnie określonymi konformacjami każde. Mac‑Diff wygenerował zaledwie 100 kandydackich struktur na białko — znacznie mniej niż typowe trajektorie symulacyjne — a mimo to odzyskał większość znanych stanów z dobrą zgodnością geometryczną. W adenylanowej kinazie wygenerował na przykład zarówno konformacje otwarte, jak i zamknięte o dużym podobieństwie do struktur krystalicznych, podczas gdy kilka popularnych metod miało skłonność faworyzować tylko jeden stan. Model działał też około tysiąc razy szybciej niż konwencjonalne symulacje na porównywalnym sprzęcie, co sprawia, że systematyczne badanie różnorodności kształtów jest znacznie bardziej praktyczne.
Co to oznacza dla biologii i medycyny
Mówiąc prosto, Mac‑Diff przekształca sekwencję białka w galerię prawdopodobnych pozy zamiast jednego portretu, robiąc to z uwzględnieniem, które części prawdopodobnie będą się stykać lub chwytać w 3D. Dzięki dokładnemu i wydajnemu próbkowaniu tych zespołów, metoda oferuje sposób na badanie, jak subtelne przesunięcia kształtu leżą u podstaw funkcji, wykrywanie rzadkich, lecz ważnych konformacji oraz poszukiwanie kieszeni wiążących leki, które pojawiają się tylko w stanach przejściowych. Chociaż nie oddaje jeszcze pełnych, uporządkowanych w czasie filmów, jakie dostarczają symulacje, Mac‑Diff udostępnia dynamiczny krajobraz białek dla wielu więcej systemów, obiecując nowe wglądy w biologię strukturalną, projektowanie leków i inżynierię białek.
Cytowanie: Wang, B., Wang, C., Chen, J. et al. Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles. Nat Mach Intell 8, 415–434 (2026). https://doi.org/10.1038/s42256-026-01198-9
Słowa kluczowe: dynamika białek, modele dyfuzyjne, zespoły konformacyjne, białka allosteryczne, odkrywanie leków