Clear Sky Science · pl
Bezprecedensowa odporność modeli energii atomowej z uwzględnieniem fizyki w temperaturach pokojowych i wyższych
Dlaczego ma to znaczenie dla codziennej chemii
Symulacje komputerowe są fundamentem współczesnej chemii i nauki o materiałach. Pozwalają naukowcom obserwować, jak cząsteczki obracają się, drgają i zderzają w środowisku cyfrowym, zamiast przeprowadzać kosztowne i czasochłonne eksperymenty. Jednak gdy symulacje opierają się na uczeniu maszynowym, mogą nagle „eksplodować”, generując nierealne kształty cząsteczek — zwłaszcza w wyższych temperaturach. W pracy tej wprowadzono nowy typ modelu uczenia maszynowego z informacją fizyczną, który potrafi prowadzić takie symulacje niezwykle długo, w temperaturach do 1000 kelwinów, bez załamania.
Od sprytnych skrótów do kruchej symulacji
Tradycyjna chemia kwantowa oblicza energie molekularne z dużą dokładnością, ale jest niezwykle wolna. Prostsze pola sił są szybkie, lecz często przybliżone. Potencjały uczone maszynowo dążą do połączenia zalet obu podejść: uczą się skrótu z geometrii molekularnej na energię i siły, a następnie używają go do napędzania dynamiki molekularnej. Na papierze wiele takich modeli wygląda świetnie, chwaląc się bardzo małymi średnimi błędami na standardowych zestawach testowych. W praktyce te liczby mogą być mylące. Gdy w trakcie symulacji cząsteczki eksplorują nowe kształty — szczególnie przy wyższych temperaturach — wiele modeli wychodzi poza zakres struktur, na których były trenowane. Zamiast łagodnie skierować cząsteczki z powrotem do realistycznych konfiguracji, mogą przewidywać siły, które rozciągają lub miażdżą wiązania do momentu, gdy cały układ staje się niefizyczny i symulacja się zawiesza.
Budowanie modeli na kwantowych blokach konstrukcyjnych
Autorzy rozwiązują tę kruchość, zmieniając to, czego model się uczy, i jak jest on prowadzony przez wcześniejszą wiedzę fizyczną. Wykorzystują ramy o nazwie FFLUX, bazujące na podejściu Interacting Quantum Atoms (IQA). W IQA cząsteczka jest podzielona na „atomy topologiczne”, których indywidualne energie są określane bezpośrednio z mechaniki kwantowej. Te energie atomowe mają sens fizyczny i sumują się do całkowitej energii cząsteczki. Zamiast uczyć arbitralnych energii miejscowych, nowe modele procesu Gaussa uczą tych kwantowo ugruntowanych energii atomowych, dostarczając głębokiego fizycznego kotwienia dla każdej prognozy. Jako wymagające testy posłużyły cztery elastyczne cząsteczki organiczne—glycyna i seryna zakończone peptydem, malondialdehyd i aspiryna—ze względu na ich liczne ruchy wewnętrzne i znane trudności dla istniejących pól sił uczonych maszynowo.
Nauczanie modelu oczekiwania problemów
Kluczową innowacją jest sposób, w jaki ustawiono proces Gaussa zanim jeszcze zobaczył dane: jego „funkcja średniej”, która koduje założenia modelu w słabo poznanych obszarach. Większość wcześniejszych prac po prostu ustawia tę średnią na zero, udając w praktyce, że model nie ma uprzednich oczekiwań. Autorzy zamiast tego celowo przesunęli tę średnią w kierunku stanów o wyższej energii atomowej, zachowując przy tym sens fizyczny. To rozwiązanie sprawia, że gdy model musi ekstrapolować — na przykład gdy wiązania są tymczasowo nadmiernie rozciągnięte — naturalnie preferuje przewidywania karzące ekstremalne deformacje. W rozległych testach wersje modelu różniące się tylko tą priorową funkcją zachowywały się diametralnie inaczej. Modele z konwencjonalnymi lub niskoenergetycznymi średnimi często przetrwały mniej niż pikosekundę, zanim cząsteczka eksplodowała lub zapadła się. Natomiast najlepsza średnia o wysokiej energii (nazwana MF5) dawała symulacje stabilne przez cały nanosekundowy okienkowy test przy temperaturach od 300 do 1000 kelwinów dla wszystkich czterech cząsteczek.
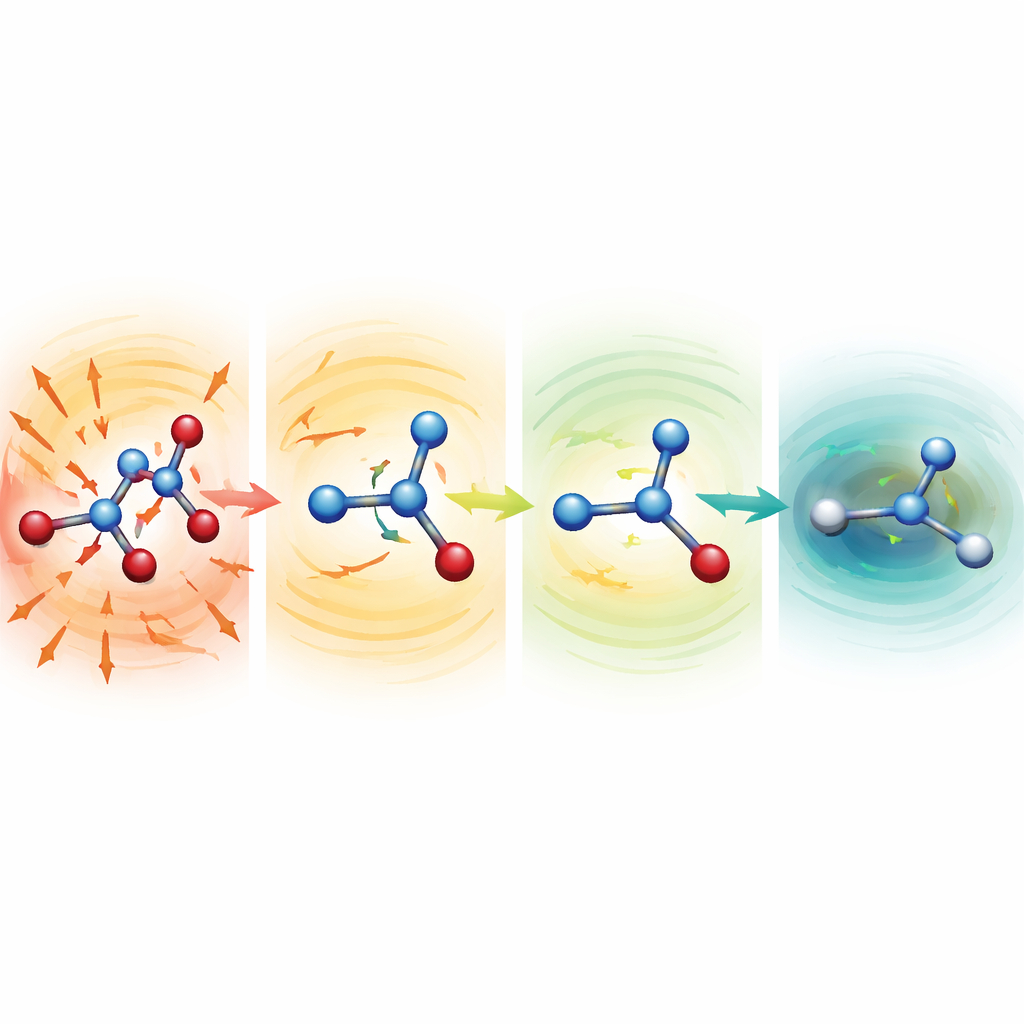
Obserwowanie, jak zdeformowane cząsteczki się rekonstruują
Aby zbadać, dlaczego odporne modele działają tak dobrze, badacze rozpoczęli symulacje od celowo zdeformowanych struktur z silnie rozciągniętymi lub ściśniętymi wiązaniami. Dla seryny, aspiryny i malondialdehydu punkty startowe leżały setki aż ponad tysiąc kilokalorii na mol powyżej normalnej struktury — konfiguracje, które normalnie byłyby katastrofalne. Przy słabszych funkcjach średniej cząsteczki szybko się rozlatywały. W konfiguracji MF5 jednak przewidywane siły natychmiast wskazywały kierunki skracające wydłużone wiązania i wydłużające ściśnięte. W ciągu dziesiątek do setek kroków czasowych cząsteczki zrelaksowały się do realistycznych kształtów, po czym dalej ewoluowały stabilnie. Zespół pokazał także, że bez treningu na siłach te same modele potrafią prowadzić optymalizacje geometryczne dipeptydu alaniny, odtwarzając znane konformacje niskoenergetyczne i względne energie z dokładnością w zakresie kilku dziesiątych kilokalorii na mol, lecz przy koszcie około 200-krotnie niższym niż pełne obliczenia kwantowe.
Długie, gorące symulacje na zwykłym sprzęcie
Odporność to nie tylko przetrwanie jednego trudnego startu; to wytrzymanie milionów lub miliardów kroków czasowych. Autorzy posunęli swoje najlepsze modele dalej, uruchamiając 50 niezależnych symulacji w 500 kelwinach, każdą przez 10 nanosekund, dla czterech testowanych cząsteczek. Żaden z tych przebiegów się nie zawiesił, dając łączny czas symulacji pół mikrosekundy — wynik rzadko spotykany wśród najnowszych pól sił uczonych maszynowo. Co więcej, symulacje działały efektywnie na standardowych procesorach CPU, krok po kroku rywalizując lub przewyższając niektóre znane potencjały oparte na sieciach neuronowych, które wymagają wydajnych GPU. W trakcie symulacji cząsteczki eksplorowały bogate zestawy kształtów i metastabilnych stanów, co pokazuje, że odporność nie została osiągnięta przez sztuczne zamrażanie ruchu czy narzucanie sztywnych struktur.
Co to oznacza dla przyszłego modelowania molekularnego
Dla osób spoza dziedziny kluczowa wiadomość jest taka, że nie wszystkie modele uczenia maszynowego o niskim błędzie są godne zaufania, gdy są mocno wystawione na próbę. Poprzez osadzenie modeli w kwantowo pochodnych energiach atomowych i staranne dostrojenie wrodzonych oczekiwań modelu w stronę stanów o wysokiej energii, autorzy stworzyli rodzinę potencjałów, które naturalnie generują „siły przywracające” — molekularny odpowiednik zabezpieczenia — utrzymujące symulacje w ramach fizycznych nawet przy wysokich temperaturach i przy zdeformowanych punktach startowych. Podejście to obiecuje bardziej wiarygodne i dłuższe symulacje złożonych cząsteczek oraz wskazuje drogę do przyszłych rozszerzeń, gdzie podobne modele uwzględniające fizykę poradzą sobie z fazami skondensowanymi i subtelnymi oddziaływaniami, takimi jak dyspersja, pozostając jednocześnie praktycznymi obliczeniowo.
Cytowanie: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Słowa kluczowe: pola sił uczenia maszynowego, odporność dynamiki molekularnej, potencjały procesu Gaussa, modelowanie uwzględniające fizykę, kwantowe energie atomowe