Clear Sky Science · pl
Dostrajanie AlphaFold przy ograniczonych obserwacjach cryo‑EM
Dlaczego kształty białek są tak trudne do zobaczenia
Białka to maleńkie molekularne maszyny, które napędzają niemal wszystkie procesy w naszych organizmach — od produkcji energii po przekazywanie impulsów nerwowych. Aby zrozumieć, jak działają i jak leki mogłyby nimi sterować, naukowcy muszą znać ich precyzyjne trójwymiarowe kształty. Do tego celu powstały dwa potężne narzędzia: cryo‑elektronowa mikroskopia (cryo‑EM), która robi wiele rozmytych zdjęć zamrożonych białek, oraz AlphaFold — system sztucznej inteligencji przewidujący struktury białek na podstawie sekwencji. W wielu rzeczywistych eksperymentach dane cryo‑EM są jednak niepełne, a przewidywania AlphaFold nie zawsze pokrywają się z rzeczywistością. W artykule opisano CoCoFold — metodę, która uczy AlphaFold słuchać bezpośrednio trudnych danych cryo‑EM i dzięki temu poprawia jego przewidywania.
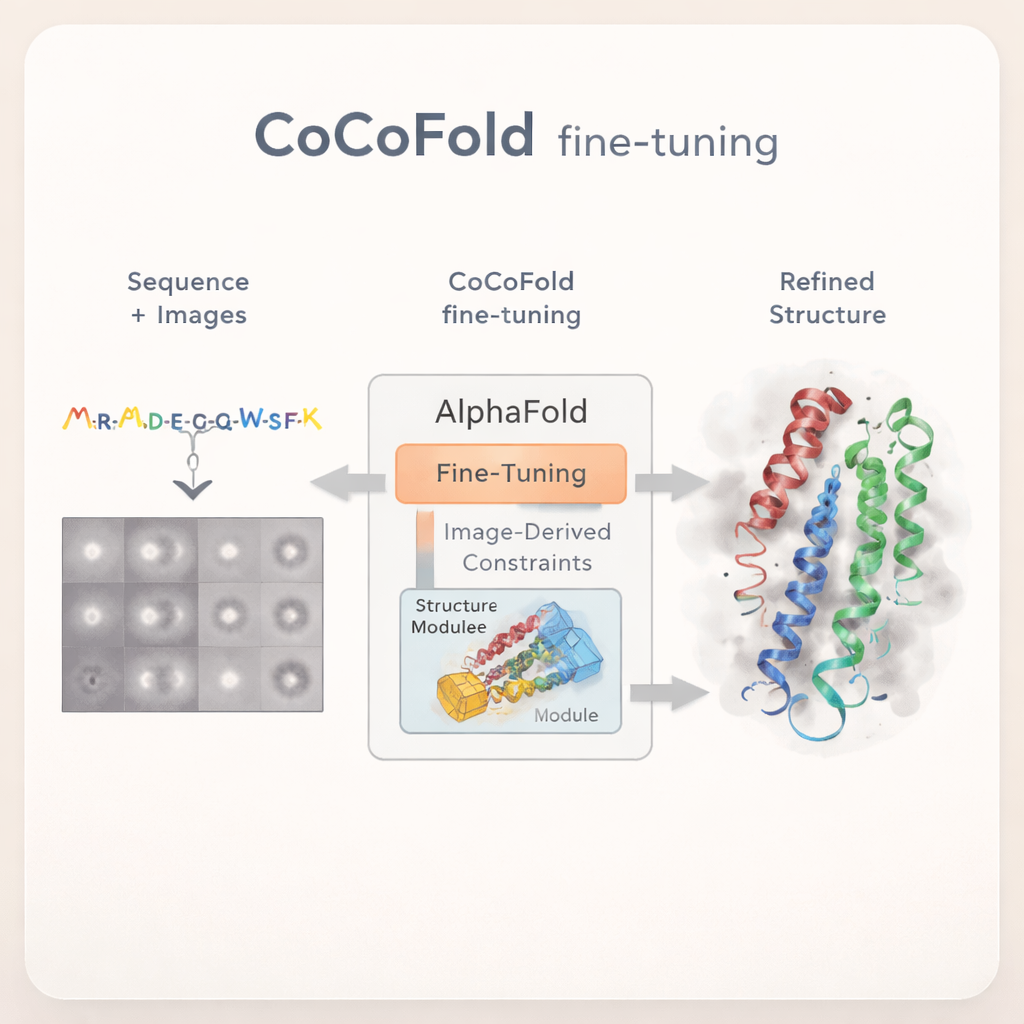
Kiedy aparat widzi za mało
Cryo‑EM polega na błyskawicznym zamrożeniu białek i uwiecznieniu ogromnej liczby pojedynczych cząstek z wielu kątów, a potem połączeniu tych obrazów w mapę 3D. W praktyce jednak badaczom często brakuje wystarczającej liczby dobrych zdjęć. Czasem białko pojawia się tylko krótko w stanie o wysokiej energii, więc uchwyconych cząstek jest bardzo niewiele. Innym razem białka preferują określone orientacje na powierzchni lodu, co powoduje brak wielu kątów widzenia. Oba problemy prowadzą do rozmytych, niepełnych map, które trudno przetłumaczyć na wiarygodne modele atomowe. Istniejące oprogramowanie potrafi dopasować przewidywane przez AlphaFold struktury do takich map, ale powodzenie zależy w dużej mierze od tego, czy na wejściu są ostre, wysokorozdzielcze dane.
Uczenie AlphaFold, by korzystał z surowych obrazów
CoCoFold proponuje inne podejście: zamiast polegać na w pełni zrekonstruowanej mapie 3D cryo‑EM, wykorzystuje bezpośrednio surowe dwuwymiarowe obrazy cząstek do dostrajania AlphaFold. Metoda zaczyna od przewidywania AlphaFold‑Multimer i utrzymuje większość pierwotnej sieci w stanie zamrożonym, zachowując jej szeroką wiedzę o fałdowaniu białek. Zmienia się jedynie końcowa część budująca strukturę. Dodany zostaje lekki „adapter”, który wprowadza do tego modułu informacje odtworzone z obrazów cryo‑EM, dyskretnie popychając model w kierunku kształtów zgodnych z danymi eksperymentalnymi, jednocześnie unikając drastycznych odchyleń od dobrze poznanej fizyki białek.
Przekształcanie obrazów w informację strukturalną
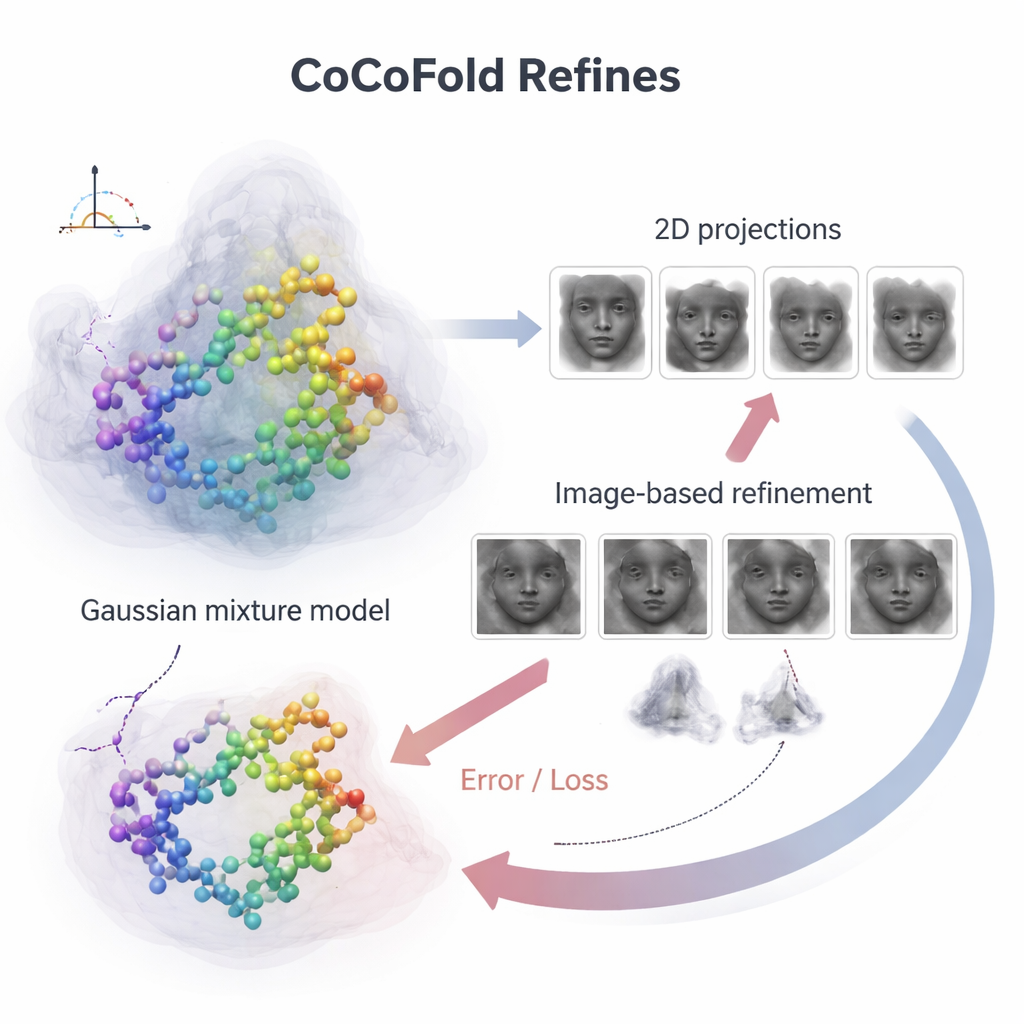
Aby powiązać poszczególne atomy białka z zaszumionymi obrazami mikroskopu, CoCoFold buduje gładki, elastyczny obraz przewidywanej struktury używając nakładających się trójwymiarowych „kropli” — mieszaniny Gaussowskiej. Z tej reprezentacji symuluje, jak białko wyglądałoby w mikroskopie przy tych samych kierunkach obserwacji i warunkach obrazowania, co w rzeczywistym eksperymencie. Takie symulowane ujęcia porównuje się następnie z rzeczywistymi cząstkami cryo‑EM, pierścień po pierścieniu w dziedzinie częstotliwości, by ocenić zgodność. Każde niedopasowanie staje się sygnałem zwrotnym, który przepływa przez sieć, lekko korygując zarówno model białka, jak i reprezentację gęstości. Po treningu model atomowy jest dodatkowo wygładzany za pomocą kroku rafinacji opartego na fizyce, aby usunąć lokalne kolizje geometryczne.
Utrzymanie dokładności przy skąpych lub obciążonych danych
Autorzy przetestowali CoCoFold na kilku eksperymentalnych i symulowanych zbiorach danych zaprojektowanych tak, by naśladować dwa główne problemy cryo‑EM: zbyt małą liczbę cząstek oraz duże luki w kątach widzenia. W tych trudnych warunkach standardowe narzędzia — w tym inne metody uczenia głębokiego zależne od zrekonstruowanych map — miały tendencję do pomijania fragmentów białka, przesuwania helis czy utraty drobnych szczegółów wraz z pogarszaniem jakości map. CoCoFold w przeciwieństwie do nich konsekwentnie tworzył modele bardziej zgodne i pełniejsze względem znanych struktur referencyjnych. Jego błędy pozostawały niewielkie nawet gdy liczba cząstek była drastycznie zmniejszona lub gdy brakowało szerokich stożków kierunków widzenia, co sugeruje, że bezpośrednie uczenie z surowych obrazów zachowuje kluczową informację, którą metody oparte na mapach wyrzucają.
Co to oznacza dla przyszłej biologii strukturalnej
Dla osób spoza specjalności najważniejszy wniosek jest taki: CoCoFold działa jak tłumacz między potężnymi przewidywaniami AI a niedoskonałymi danymi eksperymentalnymi. Zamiast polegać wyłącznie na AlphaFold lub cryo‑EM, łączy informacje z obu źródeł, zwłaszcza w trudnych sytuacjach, gdy eksperymenty dają tylko częściowy obraz. W prostych przypadkach z obfitymi, wysokiej jakości danymi narzędzia oparte na mapach wciąż działają bardzo dobrze. Jednak gdy cząstki są rzadkie lub orientacje brakują — co zdarza się często przy ściganiu krótkotrwałych lub delikatnych stanów białek — CoCoFold oferuje sposób na odzyskanie wiarygodnych modeli atomowych z informacji, które w innym wypadku poszłyby na marne.
Cytowanie: Liao, J., Zheng, D., Zhang, H. et al. Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95 (2026). https://doi.org/10.1038/s42004-026-01899-7
Słowa kluczowe: cryo‑EM, AlphaFold, struktura białka, uczenie głębokie, biologia strukturalna