Clear Sky Science · pl
Niedobór energetyczny mitochondriów leży u podstaw czuciowej neuropatii związanej z FLVCR1
Kiedy nerwy bólowe tracą zasilanie
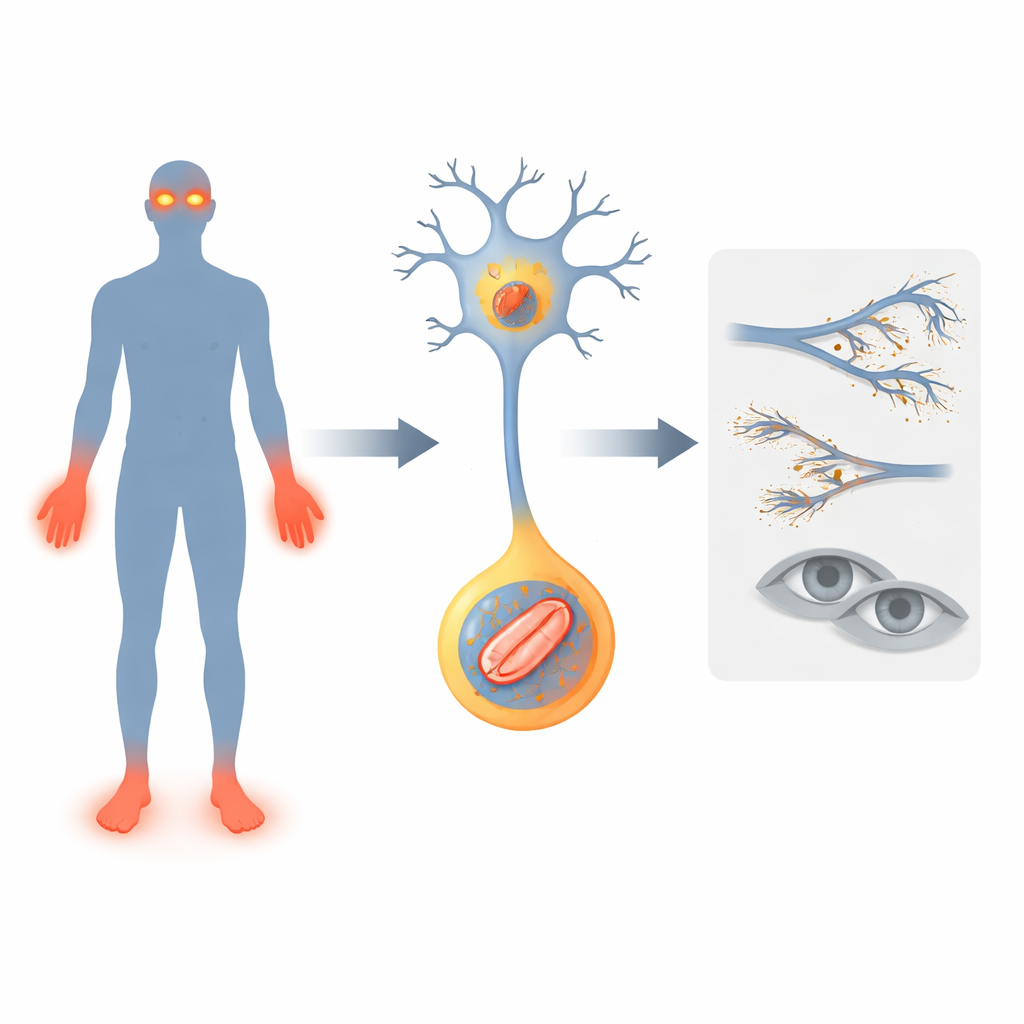
Niektórzy rodzą się niemal pozbawieni zdolności odczuwania bólu. Na pierwszy rzut oka może się to wydawać błogosławieństwem, ale szybko staje się przekleństwem: bez bólu jako sygnału ostrzegawczego gromadzą się oparzenia, złamania, zakażenia, a nawet ślepota. Badanie to analizuje rzadką postać takich zaburzeń utraty bólu i odkrywa zaskakującego winowajcę: drobne elektrownie w komórkach nerwowych, których produkcja energii zostaje poważnie zaburzona.
Gen, który ucisza alarmy
Naukowcy skupili się na genie o nazwie FLVCR1, już powiązanym z rzadkimi schorzeniami nerwowymi, w których ludzie tracą czucie bólu, rozwijają się zaburzenia chodu i czasami postępująca utrata wzroku. Opisują dwóch nowych pacjentów z wariantami w FLVCR1. Oboje dzieci wykazywały wczesne problemy: opóźnione kamienie milowe w rozwoju motorycznym, częste upadki, głębokie zakażenia i okaleczenia palców rąk i stóp, ponieważ urazy pozostawały niezauważone. U jednego rozwinęła się również zwyrodnieniowa choroba siatkówki zwana retinopatią barwnikową, prowadząca do ślepoty zmierzchowej. Przypadki te poszerzają obraz kliniczny defektów FLVCR1 i wzmacniają hipotezę, że ten gen jest kluczowy dla przetrwania nerwów czuciowych i komórek światłoczułych w siatkówce.
Modelowanie choroby u drobnych ryb
Aby zbadać, jak FLVCR1 wpływa na rozwijające się nerwy czuciowe, zespół zwrócił się ku zebrafiszom, których przezroczyste embriony umożliwiają bezpośrednią obserwację komórek nerwowych. Ograniczyli poziomy rybiej wersji genu, flvcr1a, za pomocą narzędzi genetycznych. Ryby z obniżonym flvcr1a miały mniej zwojów rdzeniowych grzbietowych, skupisk neuronów wykrywających dotyk i ból wzdłuż kręgosłupa. Behawioralnie ryby te poruszały się mniej i pływały tylko na krótkie dystanse po delikatnym dotknięciu ogona, co sugeruje stłumioną reakcję sensoryczną. Ponieważ wcześniejsze modele mysie umierały zbyt wcześnie, by przeanalizować ich nerwy czuciowe, te zebrafisze stanowią pierwszy żywy system, w którym można szczegółowo śledzić defekty nerwowe i zmiany zachowania związane z FLVCR1.
Trzy zaburzone szlaki zbiegają się przy mitochondriach
FLVCR1 znajduje się w błonach komórkowych i zarządza kilkoma kluczowymi substancjami. Wcześniejsze badania sugerowały jego rolę w gospodarce choliną (składnikiem budulcowym lipidów błonowych), hemem (barwnikiem zawierającym żelazo, niezbędnym dla wielu enzymów) oraz w przepływie wapnia między przedziałami komórkowymi. Naukowcy zebrali fibroblasty skóry od czterech pacjentów z różnymi mutacjami FLVCR1 i porównali je z komórkami od zdrowych osób i bezobjawowych nosicieli. Stwierdzili, że komórki pacjentów miały niższe stężenia choliny i bardziej płynne błony komórkowe — zmiany, które mogą zaburzać delikatne środowisko lipidowe wymagane przez mitochondria, organelle wytwarzające energię. Odkryli również, że kluczowy enzym syntezy hemu w mitochondriach, ALAS1, był mniej aktywny, mimo że całkowita zawartość hemu wydawała się prawie normalna. Jednocześnie miejsca kontaktu między siateczką śródplazmatyczną a mitochondriami — gdzie zwykle przepływa wapń do mitochondriów — były krótsze i rzadsze, a napływ wapnia do mitochondriów był zmniejszony. Trzy problemy — niedobór choliny, spowolniona produkcja hemu i osłabiony transfer wapnia — wszystkie wskazywały na pogorszoną wydajność mitochondriów.
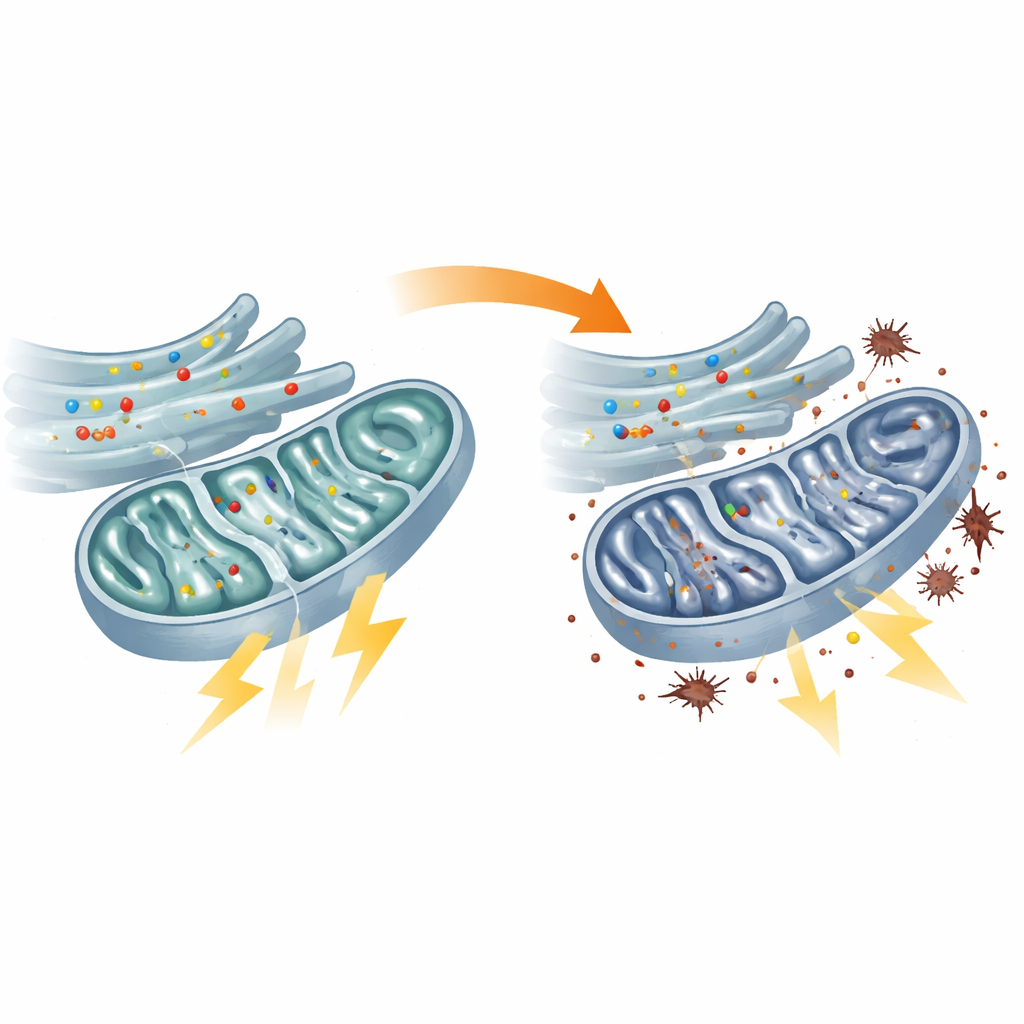
Bezpośrednie testy metabolizmu energetycznego potwierdziły, że mitochondria w fibroblastach pacjentów działały słabiej. Centralny szlak przetwarzania paliwa, znany jako cykl TCA, przebiegał wolniej, kilka jego kluczowych enzymów miało niższą aktywność, a łańcuch reakcji prowadzący do produkcji ATP, waluty energetycznej komórki, był przytłumiony. W rezultacie poziomy ATP w mitochondriach spadły. Komórki próbowały to zrekompensować poprzez zwiększenie glikolizy, mniej wydajnej ścieżki spalania cukrów poza mitochondriami. Ta zmiana strategii energetycznej miała cenę: elektrony wyciekające z przeciążonego sprzętu mitochondrialnego wywołały wyższe poziomy peroksydacji lipidów, formy uszkodzenia oksydacyjnego błon komórkowych. Podobne defekty zaobserwowano u zebrafiszy z obniżonym flvcr1a, łącząc niewydolność mitochondriów bezpośrednio z modelem zwierzęcym czuciowej neuropatii.
Wskazówki dotyczące przyszłych terapii przez wzmocnienie energii komórkowej
Co ważne, niektóre z tych defektów dało się złagodzić w laboratorium. Gdy zespół sztucznie zwiększył napływ wapnia do mitochondriów przez nadprodukcję kanału białkowego MCU w komórkach pacjentów, produkcja energii odbiła się, a oznaki uszkodzeń oksydacyjnych zmalały. Dostarczanie komórkom prekursora hemu, kwasu 5-aminolewulinowego (ALA), również poprawiło aktywność cyklu TCA, funkcję łańcucha oddechowego i poziomy ATP, chociaż długotrwała ekspozycja na ALA była szkodliwa w poprzednich badaniach. Dodatkowa cholina unormowała płynność błon i pomogła zmniejszyć uszkodzenia lipidów, ale przyniosła tylko skromne, krótkotrwałe korzyści w produkcji energii. Eksperymenty ratunkowe sugerują, że żaden pojedynczy szlak nie jest wyłącznym winowajcą; zamiast tego sieć zaburzeń w gospodarce choliną, hemem i wapniem popycha mitochondria w stan chronicznej niewydolności.
Dlaczego te ustalenia mają znaczenie dla pacjentów
Śledząc konsekwencje mutacji FLVCR1 od cząsteczek, przez komórki, aż po całe organizmy, praca ta proponuje, że niewydolność energetyczna mitochondriów jest siłą napędową tej postaci neuropatii utraty bólu oraz związanych z nią problemów ze wzrokiem. Nerwy czuciowe i fotoreceptory mają wyjątkowo duże zapotrzebowanie na energię, ponieważ utrzymują długie aksony lub nieustannie odnawiają struktury światłoczułe, co czyni je szczególnie wrażliwymi, gdy wydajność mitochondriów spada. Model zebrafiszy i komórki pochodzące od pacjentów oferują teraz praktyczne pole do testowania terapii wzmacniających metabolizm mitochondrialny. Choć leczenia takie jak suplementacja choliny, kontrolowane zwiększanie hemu czy leki zwiększające pobór wapnia przez mitochondria będą wymagały ostrożnej oceny w modelach zwierzęcych i badaniach klinicznych, główne przesłanie jest jasne: przywrócenie zasilania delikatnych neuronów może pewnego dnia pomóc chronić osoby urodzone bez najważniejszego sygnału ostrzegawczego od natury — bólu.
Cytowanie: Bertino, F., Zanin Venturini, D.I., Grasso, E. et al. Mitochondrial energetic failure underlies FLVCR1-related sensory neuropathy. Commun Biol 9, 429 (2026). https://doi.org/10.1038/s42003-026-09691-y
Słowa kluczowe: neuropatia czuciowa, dysfunkcja mitochondriów, FLVCR1, niewrażliwość na ból, metabolizm energetyczny nerwów