Clear Sky Science · pl
haCCA: wielomodułowa integracja przestrzennych transkryptomów i metabolomów opartych na punktach
Dlaczego ważne jest mapowanie cząsteczek w miejscu
Nasze ciała zbudowane są z niezliczonych małych sąsiedztw komórek, z których każde ma własny zestaw aktywnych genów i substancji chemicznych. Jeszcze niedawno naukowcy musieli badać te molekuły po zmieleniu tkanki na jednorodną masę, tracąc całkowicie informację o tym, „gdzie” się znajdowały. W tym artykule przedstawiono nową metodę obliczeniową, nazwaną haCCA, która łączy dwie mocne techniki obrazowania, pozwalając badaczom zobaczyć in situ, jak geny i małe cząsteczki rozmieszczone są w rzeczywistych tkankach i guzach. Tego rodzaju mapa może ujawnić ukryte wzorce chorobowe i zasugerować bardziej precyzyjne terapie.
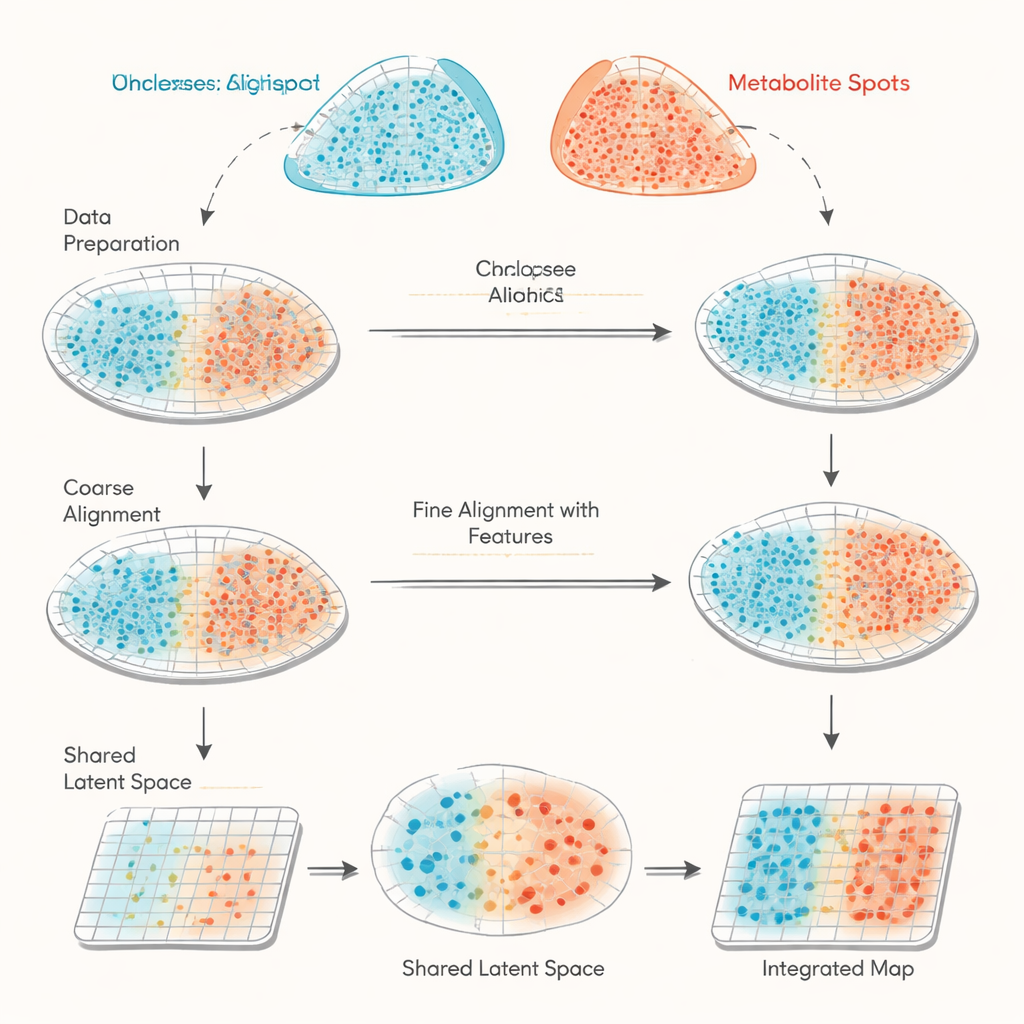
Dwa różne spojrzenia na tę samą tkankę
Badanie skupia się na łączeniu danych z dwóch metod przestrzennych, które są coraz częściej używane w biologii. Przestrzenna transkryptomika rejestruje, które geny są włączone w tysiącach drobnych punktów na plaszczie tkanki. Obrazowanie masowe MALDI (MALDI mass spectrometry imaging) mierzy ilości wielu małych cząsteczek, takich jak metabolity i lipidy, na podobnie gęstych siatkach punktów. Problem polega na tym, że oba instrumenty nie mierzą dokładnie tych samych pozycji ani tego samego zestawu cech, więc ich dane przypominają dwie źle wyrównane mapy z różnymi legendami. Istniejące podejścia w dużej mierze starają się dopasować kształty przekrojów tkanki bazując wyłącznie na współrzędnych, co może być niedokładne i nie daje sposobu na sprawdzenie, jak dobrze wyrównanie faktycznie zadziałało.
Mądrzejszy sposób wyrównywania map molekularnych
haCCA (skrót od hierarchical anchor-guided canonical correlation analysis) rozwiązuje to wyzwanie, łącząc geometrię z biologią. Najpierw wykonuje dwuetapowe „wyrównanie morfologiczne” siatek punktów z obu technologii. Eksperci ludzie wybierają kilka odpowiadających sobie punktów orientacyjnych na obrazach tkanki, aby w przybliżeniu skorygować przesunięcia i rotacje, a następnie krok automatyczny dopracowuje odchylenia przy rozdartych krawędziach lub brakujących fragmentach. Następnie metoda wyszukuje „kotwice” — pary punktów, które są blisko siebie w przestrzeni i leżą w lokalnie jednorodnych obszarach, co zwiększa prawdopodobieństwo, że reprezentują ten sam region tkanki. Na podstawie tych punktów kotwiczych haCCA oblicza, które geny i metabolity mają tendencję do współzmienności, i destyluje je do wspólnej, niskowymiarowej reprezentacji, która wychwytuje najsilniejsze wspólne wzorce.
Przekształcanie korelacji w jednolity obraz tkanki
Mając zarówno współrzędne przestrzenne, jak i wspólną reprezentację molekularną, haCCA rozwiązuje problem optymalizacyjny, aby określić, jak prawdopodobne jest dopasowanie każdego punktu genowego do każdego punktu metabolitowego. Ten etap jest zaprojektowany tak, by utrzymywać punkty blisko w przestrzeni, ale także podobne pod względem ich łącznego profilu gen–metabolit. Końcowym rezultatem jest „plan transportu”, który łączy każdy punkt w jednym zestawie danych z jego najlepszym partnerem w drugim, tworząc zintegrowaną mapę multimodalną. Na starannie skonstruowanych danych testowych — gdzie prawdziwe relacje są znane — autorzy pokazują, że każdy etap przepływu pracy (wstępne wyrównanie, dopracowane wyrównanie i dopasowanie uwzględniające cechy) stopniowo poprawia trzy niezależne miary dokładności. W porównaniu z innymi narzędziami opierającymi się głównie na geometrii, haCCA konsekwentnie osiąga lepsze wyrównanie i wierniejsze przenoszenie etykiet regionów.
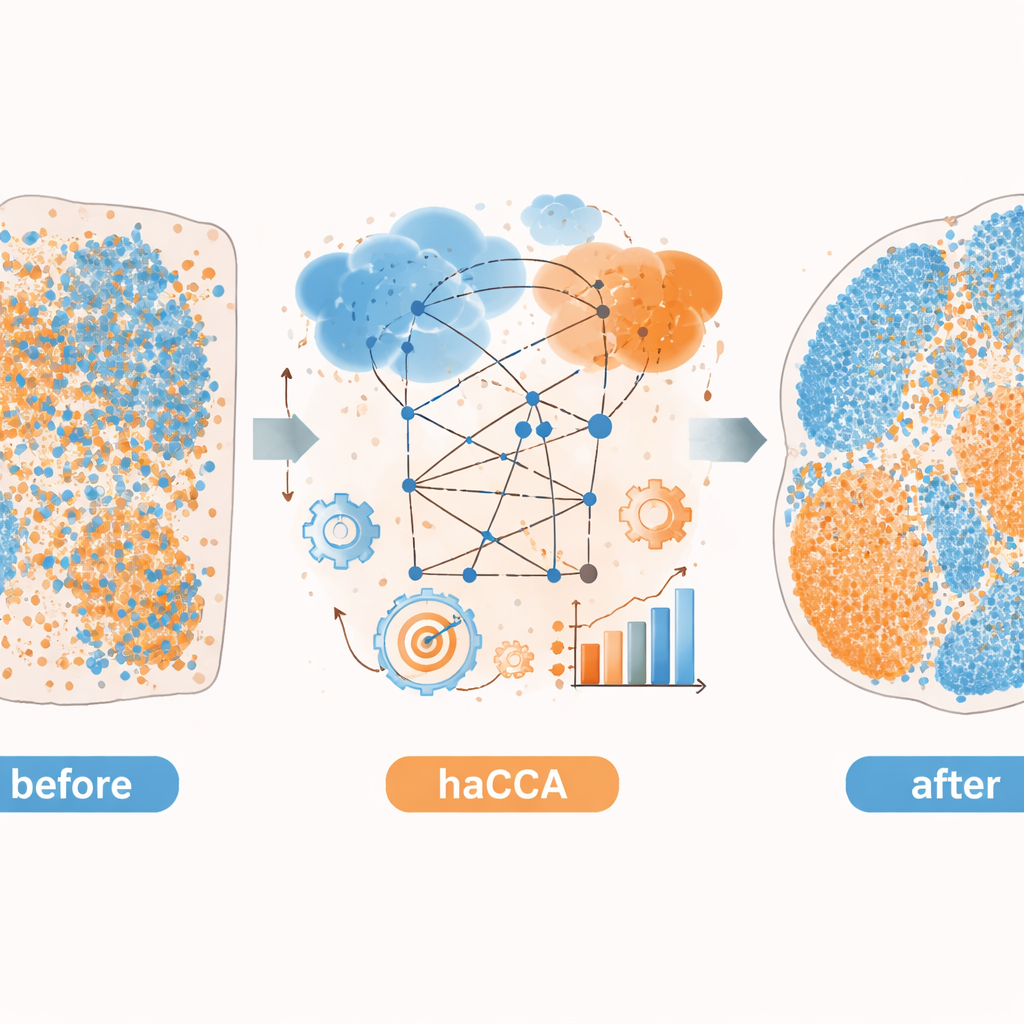
Odkrywanie ukrytej biologii w nowotworach mózgu i wątroby
Następnie autorzy zastosowali haCCA do rzeczywistych próbek tkanki mózgu myszy oraz guzów wątrobowych. Dla mózgu zintegrowali komercyjne dane przestrzennej transkryptomiki z obrazami metabolitów pochodzącymi z tej samej lub sąsiedniej sekcji. Metoda zachowuje znane terytoria metaboliczne i rekonstruuje oczekiwane nakładania się, takie jak współlokalizacja dopaminy z genem kodującym jej kluczowy enzym. Poprzez wspólne grupowanie (clustering) genów i metabolitów odkryli, że połączone dane rozróżniają bardziej subtelne podregiony tkanki niż każda z modalności osobno. W modelu przedklinicznym wewnątrzwątrobowego raka dróg żółciowych (intrahepatic cholangiocarcinoma), typu nowotworu wątroby, użyli haCCA do porównania guzów, które potrafią lub nie potrafią tworzyć neutrofilowych pułapek zewnątrzkomórkowych — sieci uwalnianych przez komórki odpornościowe. Zintegrowane mapy wykazały, że gdy te pułapki są obecne, gen zwany Scd1 i powiązane z nim kwasy tłuszczowe są wzbogacone w obszarach złośliwych, co wskazuje na przesunięcie w kierunku zmienionego metabolizmu lipidów w guzie.
Co to oznacza dla przyszłych badań
Mówiąc prosto, haCCA przypomina wyrównywanie zdjęć lotniczych wykonanych różnymi kamerami — jedna czuła na zarysy budynków, druga na sygnatury cieplne — aby uzyskać ostrzejszy obraz tego, co dzieje się w każdym bloku miasta. Dzięki dokładnemu łączeniu informacji o tym, gdzie geny są aktywne, z miejscami akumulacji kluczowych metabolitów, ten przepływ pracy pomaga naukowcom jednocześnie profilować obie strony zachowania komórek: instrukcje i wynikającą z nich chemię. Podejście ulepsza wcześniejsze metody wyrównywania, jest udostępnione w przystępnym narzędziu w Pythonie i można je rozszerzyć na inne technologie przestrzenne. W miarę jak takie zintegrowane mapy staną się powszechniejsze, mogą pogłębić nasze zrozumienie, jak guzy i inne tkanki organizują swój metabolizm, reagują na leczenie i ewoluują w czasie.
Cytowanie: Xu, J., Shen, XT., Zhang, C. et al. haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes. Commun Biol 9, 248 (2026). https://doi.org/10.1038/s42003-026-09526-w
Słowa kluczowe: przestrzenne multi-omiki, transkryptomika, metabolomika, metabolizm nowotworu, integracja danych