Clear Sky Science · pl
Oś sygnalizacyjna AXL–SHC1 pośredniczy w adaptacyjnej oporności na inhibitory kinazy tyrozynowej ukierunkowane na HER2 w nowotworach płuca i żołądka z aberracjami HER2
Dlaczego niektóre nowotwory przechytrzają nowe leki
Leki celowane w nowotwory zrewolucjonizowały leczenie wielu pacjentów, koncentrując się na określonych cząsteczkach napędzających wzrost guza. Nawet przy tych precyzyjnych terapiach całkowite i trwałe remisje pozostają jednak rzadkie. W badaniu postawiono pilne pytanie dotyczące pacjentów z rakiem płuca i żołądka napędzanym przez gen HER2: dlaczego guzy, które początkowo kurczą się po lekach blokujących HER2, niemal zawsze pozostawiają uporczywe jądro komórek, które później napędza nawroty — i jak lekarze mogliby zablokować tę drogę ucieczki od samego początku?
Bliższe spojrzenie na guzy zależne od HER2
HER2 jest węzłem sygnalizacyjnym wspierającym wzrost i przeżycie komórek. Gdy jest zmieniony lub nadmiernie eksprymowany, może przekształcić normalne komórki w nowotworowe w piersi, żołądku lub płucu. Kilka nowoczesnych leków z grupy inhibitorów kinaz tyrozynowych (TKI) zostało zaprojektowanych, by wyłączać HER2 wewnątrz komórek nowotworowych. Leki te, w tym mobocertynib i inne, mogą zmniejszać rozmiar guzów i opóźniać progresję choroby. W raku płuca i żołądka ich korzyści są jednak często tymczasowe. Niewielka frakcja komórek guza potrafi przetrwać początkowy atak lekowy w stanie tolerancji na lek, a następnie ewoluować w w pełni oporne guzy. Zrozumienie, co utrzymuje przy życiu te przetrwałe komórki, jest kluczowe do zaprojektowania mądrzejszych terapii pierwszego rzutu.
Zapasowa droga ratunkowa zwana AXL
Naukowcy przesiewali linie komórkowe raka płuca i żołądka z aberracjami HER2, aby sprawdzić, które inne przełączniki sygnalizacyjne pomagają komórkom przetrwać leczenie TKI ukierunkowanymi na HER2. Zidentyfikowali receptor o nazwie AXL jako kluczowego gracza. Po ekspozycji na leki blokujące HER2 AXL stawał się aktywowany i pozostawał włączony, nawet gdy główne sygnały zależne od HER2 były osłabione. Ta aktywacja podtrzymywała ważną ścieżkę przetrwania znaną jako AKT–mTOR. Wyłączenie AXL przy użyciu narzędzi genetycznych lub zablokowanie go eksperymentalnymi lekami sprawiało, że komórki nowotworowe stawały się znacznie bardziej wrażliwe na różne TKI ukierunkowane na HER2, prowadząc do zmniejszenia wzrostu komórek i zwiększonej śmierci komórkowej w warunkach laboratoryjnych.
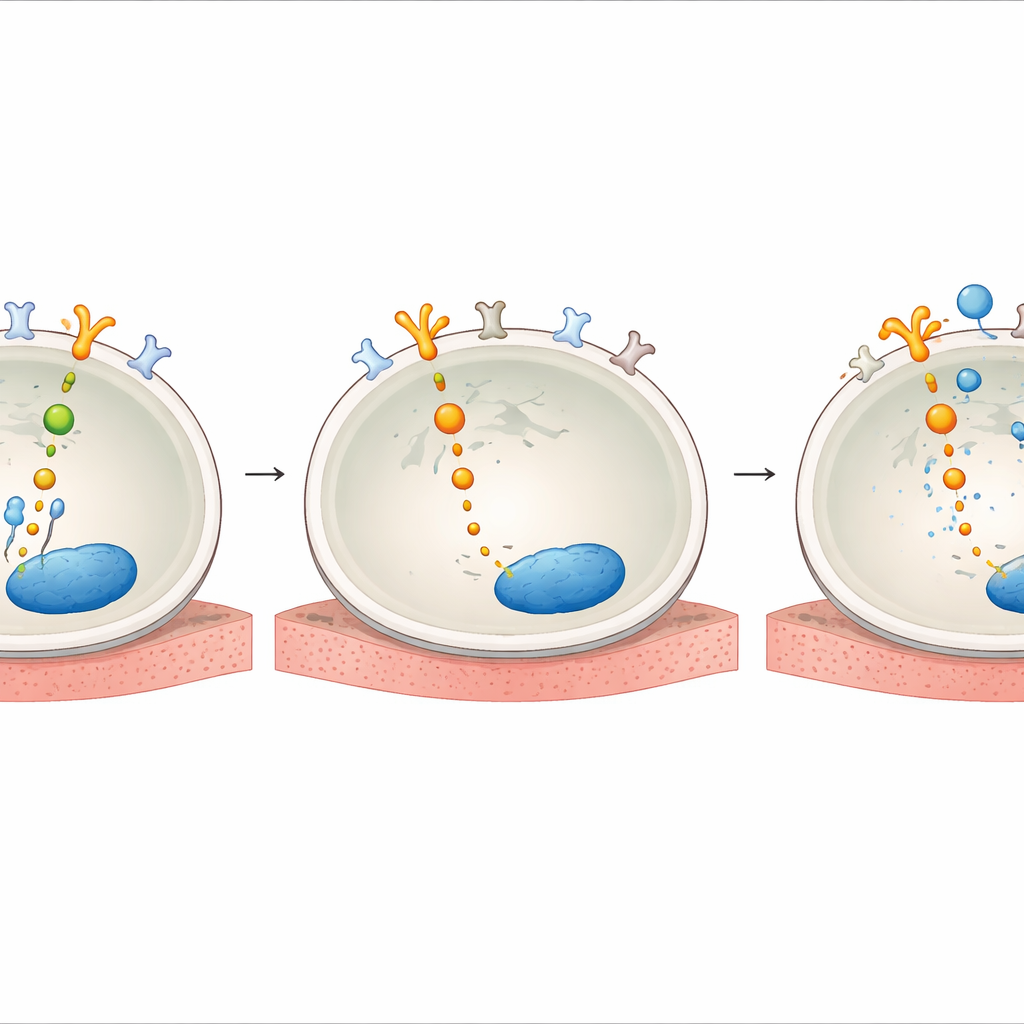
Jak białka pomocnicze organizują drogę ucieczki
Zespół badał następnie, w jaki sposób AXL zyskuje taką przewagę pod presją leku. Stwierdzili, że leczenie TKI przeciw HER2 zwiększa poziomy GAS6, naturalnego liganda aktywującego AXL na powierzchni komórki. Pokazali też, że po ekspozycji na lek AXL fizycznie łączy się z HER2 i jego rodzeństwem EGFR oraz HER3, efektywnie podłączając się do tej samej logiki przetrwania, którą normalnie kontroluje HER2. Wewnątrz komórki białka adapterowe nazwane SHC1 i SHCBP1 działały jak węzły okablowania. Gdy HER2 został zablokowany, SHC1 odłączał się od SHCBP1 i zamiast tego wiązał się z AXL, podczas gdy SHCBP1 przemieszczał się do jądra, gdzie pomagał napędzać cykl komórkowy. Redukcja ekspresji SHC1 lub SHCBP1 osłabiała sygnalizację AKT i zmniejszała przeżywalność komórek, ujawniając oś AXL–SHC1–SHCBP1, która podtrzymuje wzrost, gdy HER2 jest zahamowany.
Powstrzymanie komórek tolerujących lek zanim się utrwalą
Aby naśladować to, co dzieje się u pacjentów, naukowcy pozwolili komórkom nowotworowym rosnąć przez kilka dni w obecności mobocertynibu, wybierając małą populację, która tolerowała lek. Te komórki tolerujące lek rosły wolniej, ale były wyraźnie mniej wrażliwe na inhibitor HER2 niż komórki pierwotne. Ich przeżycie nadal w dużej mierze zależało od AXL: dodanie inhibitora AXL ostro ograniczało ich wzrost i tłumiło aktywność AKT. W modelach mysi z guzami płuca z aberracją HER2 i dodatnim oznakowaniem AXL, łączenie mobocertynibu z inhibitorem AXL zmniejszało guzy bardziej, redukowało liczbę komórek dzielących się i zwiększało oznaki zaprogramowanej śmierci komórek w porównaniu z samym lekiem przeciw HER2 — bez dodatkowej toksyczności. Guzy zmodyfikowane do nadprodukcji AXL były wyraźnie mniej wrażliwe na sam mobocertynib, ale ponownie reagowały, gdy dodano inhibitor AXL.
Co to może znaczyć dla przyszłego leczenia
Co ważne, próbki tkanek od pacjentów z rakiem płuca i żołądka z aberracjami HER2 wykazały, że około jedna czwarta miała wysokie poziomy AXL, a większość miała przynajmniej pewien jego poziom. Sugeruje to, że znacząca grupa pacjentów mogłaby odnieść korzyść ze strategii leczenia łączącej TKI ukierunkowany na HER2 z inhibitorem AXL od pierwszego dnia, zamiast czekać na pojawienie się oporności. Mówiąc prosto, badanie pokazuje, że wiele guzów zależnych od HER2 utrzymuje zapasowy przełącznik wzrostu — AXL — w stanie gotowości. Gdy HER2 jest zablokowany, AXL przejmuje dowodzenie i podtrzymuje życie komórek nowotworowych. Wyłączenie obu przełączników jednocześnie powoduje znacznie większą śmierć komórek nowotworowych i spowalnia lub zapobiega powstawaniu komórek tolerujących lek. Jeśli potwierdzą to badania kliniczne, takie podejście podwójnego celu mogłoby prowadzić do dłużej utrzymującej się kontroli nad rakiem płuca i żołądka z aberracjami HER2.
Cytowanie: Ishida, M., Yamada, T., Katayama, Y. et al. AXL–SHC1 signaling axis mediates adaptive resistance to HER2-targeted tyrosine kinase inhibitors in HER2-aberrant lung and gastric cancers. npj Precis. Onc. 10, 142 (2026). https://doi.org/10.1038/s41698-026-01385-2
Słowa kluczowe: terapia ukierunkowana na HER2, inhibitor AXL, oporność na leki, rak płuca, rak żołądka