Clear Sky Science · pl
Odpowiedź na midostaurynę w AML kształtowana przez stan komórkowy przypominający progenitor, selektywnie atakowany przez mimetyki SMAC
Dlaczego niektóre leki przeciw białaczce przestają działać
Dla wielu osób z rodzajem nowotworu krwi zwanego ostrą białaczką szpikową (AML) nowe leki ukierunkowane przyniosły nadzieję — ale nie wszyscy odnoszą korzyść, a odpowiedzi często słabną. W tym badaniu postawiono proste, lecz kluczowe pytanie: dlaczego niektóre komórki białaczkowe ignorują powszechnie stosowany lek midostaurynę i czy można znaleźć inteligentne połączenie, które zmusi te oporne komórki do śmierci?
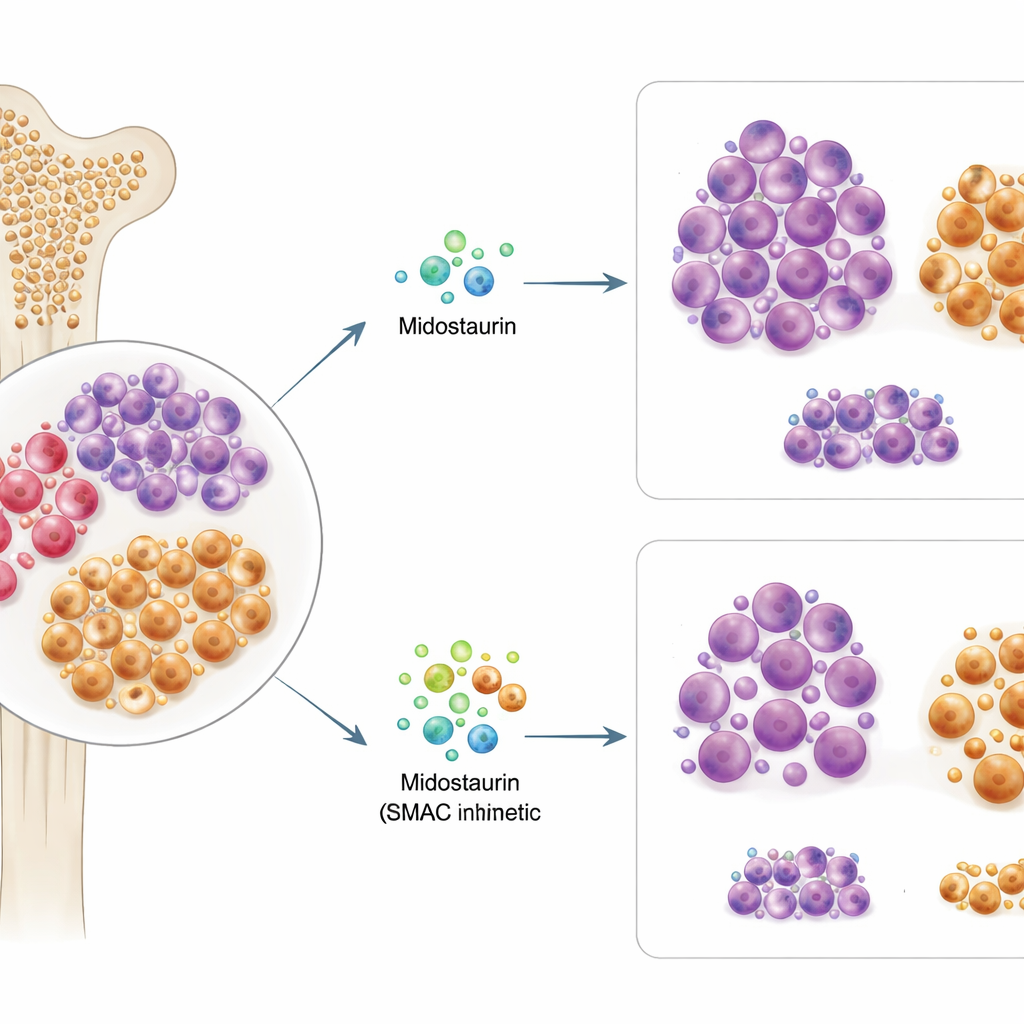
Patrząc poza główną mutację
Około u jednej na trzy osoby z AML występuje zmiana w genie FLT3, która napędza rozwój białaczki i jest powodem stosowania midostauryny. Naukowcy przebadali próbki szpiku kostnego i krwi od 63 pacjentów z AML z mutacją FLT3, wystawiając komórki na działanie midostauryny i ponad 500 innych leków przeciwnowotworowych in vitro. Stwierdzili, że stopień wrażliwości komórek pacjenta na midostaurynę ex vivo ściśle korelował z późniejszą odpowiedzią kliniczną tego pacjenta. Zaskakująco, dokładny typ mutacji FLT3 ani jej obfitość nie przewidywały wiarygodnie skuteczności midostauryny, co sugeruje, że sama genetyka nie wyjaśnia, kto odniesie korzyść.
Ukryta pula opornych „nasion”
Zagłębiając się bardziej, zespół porównał całkowite wzorce białkowe i ekspresję genów między komórkami wrażliwymi na midostaurynę a tymi niewrażliwymi. Próbki niewykazujące odpowiedzi były wzbogacone w cechy niedojrzałych, przypominających komórki macierzyste progenitorów — komórek bliższych korzeniowi tworzenia krwi, które uważa się za „nasiona” zdolne do ponownego wznowienia białaczki. Natomiast próbki reagujące przypominały bardziej częściowo zróżnicowane komórki odpornościowe i mieloidalne. Przy użyciu zaawansowanych metod jednopkomórkowych naukowcy zidentyfikowali specyficzną populację komórek białaczkowych oznakowanych białkami powierzchniowymi CD38 i CD45RA, które zachowywały się jak te progenitoropodobne nasiona. Komórki te wykazywały nietypową organizację błony zewnętrznej, co sugeruje, że kluczowe cząsteczki sygnałowe były ustawione w sposób sprzyjający przetrwaniu.
Okablowanie przetrwania: przełączenie szlaków sygnałowych
Midostauryna ma blokować sygnalizację FLT3, która zwykle uruchamia kaskadę sygnałów obejmującą między innymi cząsteczkę STAT5 i może napędzać wzrost komórek. Gdy zespół badał sygnalizację w liniach komórkowych i próbkach pacjentów po leczeniu midostauryną, zaobserwowano dwa wyraźne wzory. W komórkach wrażliwych aktywność STAT5 szybko spadała, co odpowiada skutecznemu wyłączeniu FLT3. W komórkach opornych dominował jednak inny szlak: PI3K/AKT, klasyczna ścieżka przetrwania, która pomaga komórkom przeciwstawiać się śmierci. Komórki oporne utrzymywały lub nawet zwiększały aktywność AKT po leczeniu i wykazywały wyższe poziomy białek blokujących apoptozę (zaprogramowaną śmierć komórki). Innymi słowy, we wnętrzu tych progenitoropodobnych komórek okablowanie sygnałowe wydawało się przekierowane tak, by faworyzować przetrwanie nawet przy zahamowanym FLT3.
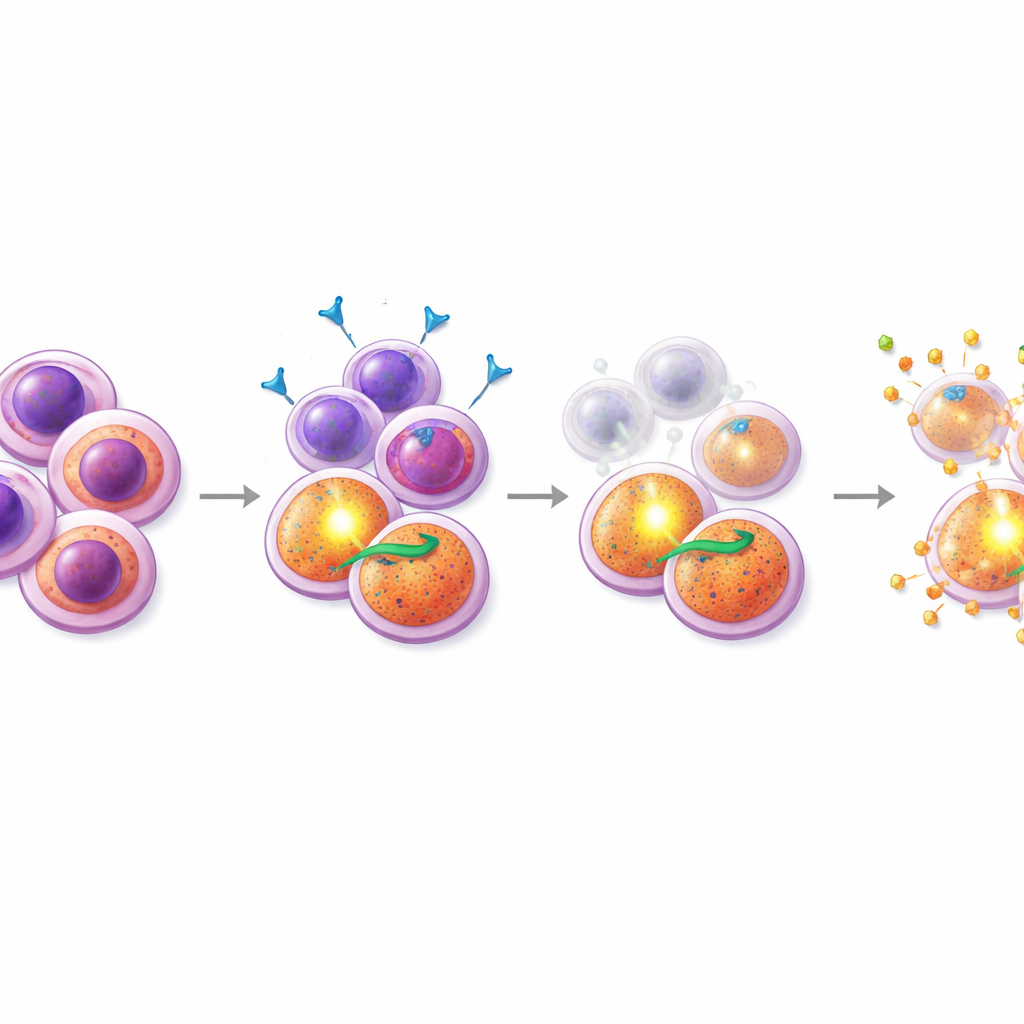
Znajdowanie partnera, który uderza w słaby punkt
Wyposażeni w tę wiedzę, badacze przetestowali kombinacje midostauryny z setkami innych związków, skupiając się na lekach wpływających na śmierć komórek. Jedną z wyróżniających się grup okazały się mimetyki SMAC, leki unieszkodliwiające białka "inhibitorów apoptozy", na których polegają komórki napędzane przez PI3K/AKT. W próbkach opornych pacjentów i w opornej linii komórkowej z mutacją FLT3 dodanie mimetyków SMAC, takich jak birinapant, do midostauryny dało silną synergię: w połączeniu leki zabijały znacznie więcej komórek niż każdy z nich osobno. Co istotne, szczegółowe eksperymenty z cytometrią przepływową wykazały, że kombinacja midostauryna–mimetyk SMAC selektywnie wyczerpywała populację progenitoropodobnych komórek CD38+CD45RA+ i obniżała poziomy ich charakterystycznych markerów powierzchniowych, sugerując, że ta terapia specyficznie trafia w trudno zabijalne nasiona. Dla porównania, kombinacje z zatwierdzonym inhibitorem BCL-2, wenetoklaksem, były skuteczniejsze wobec innej podgrupy z wysokim poziomem CD34 i nie wykazywały tego samego ukierunkowanego efektu na komórki oporne.
Co to oznacza dla pacjentów
Ta praca sugeruje, że oporność na midostaurynę to nie tylko kwestia samej mutacji FLT3, lecz także „stanu” komórek białaczkowych — ich stopnia dojrzałości, organizacji błony i preferowanych dróg przetrwania. Progenitoropodobna podpopulacja CD38+CD45RA+ wydaje się być kluczowym rezerwuarem oporności, przełączając sygnalizację z typowej ścieżki STAT5 na korzystny dla przetrwania program PI3K/AKT. Łącząc midostaurynę z mimetykami SMAC, badacze byli w stanie ponownie uczulić te komórki i sprowokować ich śmierć w badaniach laboratoryjnych. Chociaż potrzebne są większe badania kliniczne, wyniki wskazują na przyszłość, w której lekarze mogą używać testów funkcjonalnych i profilowania stanu komórek, nie tylko sekwencjonowania DNA, aby dobierać kombinacje ukierunkowane na FLT3, które wyeliminują zarówno masę białaczki, jak i jej najbardziej wytrwałe nasiona.
Cytowanie: Struyf, N., Gezelius, H., Lundmark, A. et al. Midostaurin response in AML is shaped by a progenitor-like cell state selectively targeted by SMAC mimetics. npj Precis. Onc. 10, 117 (2026). https://doi.org/10.1038/s41698-026-01363-8
Słowa kluczowe: ostra białaczka szpikowa, inhibitory FLT3, oporność na leki, komórki macierzyste białaczki, mimetyki SMAC