Clear Sky Science · pl
Ab initio określenie stabilności faz dynamicznie nieuporządkowanych ciał stałych: rotacyjne nieuporządkowanie C2 w Li2C2
Dlaczego ten zmieniający się materiał ma znaczenie
Wiele współczesnych technologii opiera się na ciałach stałych, które potrafią w sposób niemal niezauważalny zmieniać swoją wewnętrzną strukturę pod wpływem ogrzewania lub ściskania. Zmiany te, zwane przejściami fazowymi, są kluczowe dla koncepcji takich jak chłodzenie w stanie stałym czy bezpieczniejsze ogniwa. W tej pracy badano prosty związek, węglik litu (Li2C2), który wraz ze wzrostem temperatury przechodzi z dobrze uporządkowanej formy do bardziej ruchliwej, dynamicznie nieuporządkowanej postaci. Obserwując tę przemianę atom po atomie w symulacjach komputerowych, autorzy pokazują, jak wewnętrzna „nerwowość” małych jednostek cząsteczkowych może przesunąć równowagę między dwiema strukturami krystalicznymi.
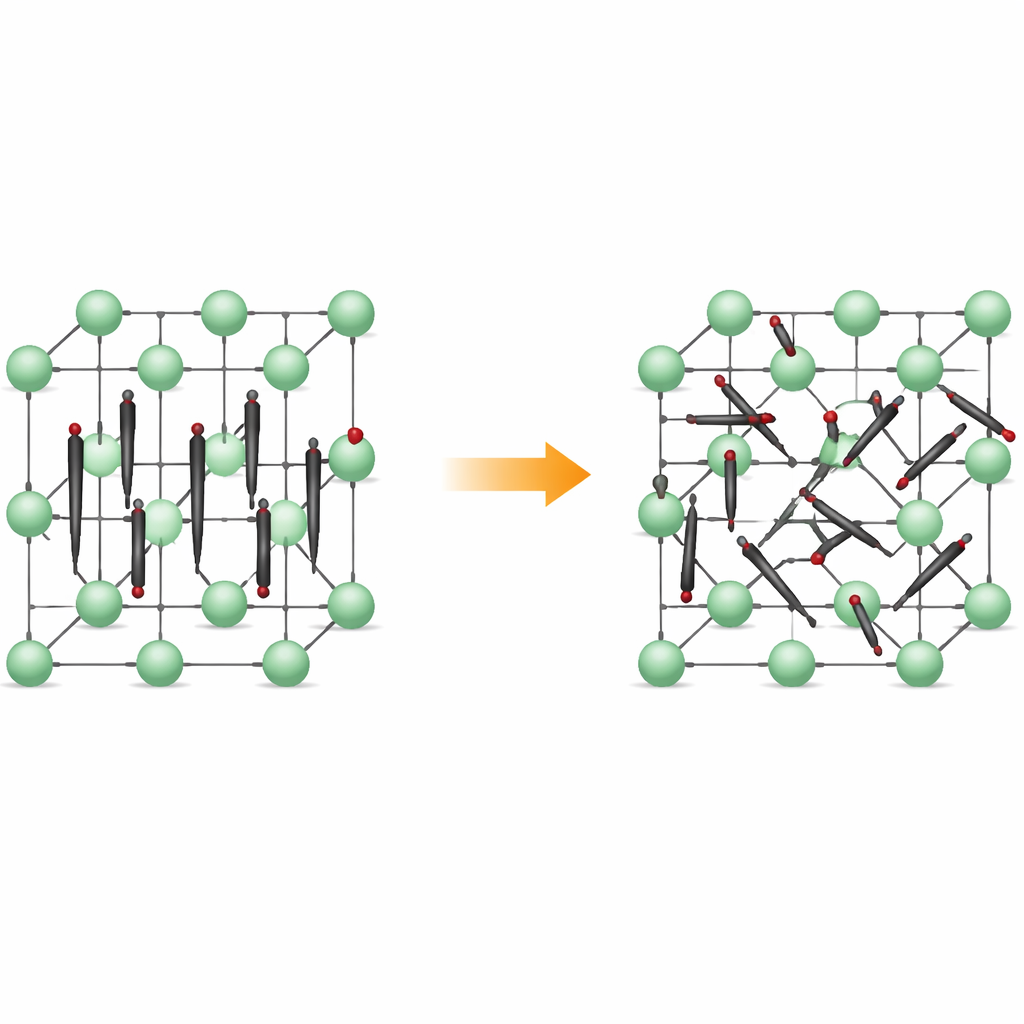
Od uporządkowanych rzędów do nieustannego ruchu
W niskich temperaturach Li2C2 tworzy kryształ o strukturze ortorombicznej: atomy węgla łączą się w pary C2, które wszystkie wskazują w niemal tym samym kierunku, niczym wyrównane zapałki. Jony litu osadzają się pomiędzy nimi, tworząc regularną, trójwymiarową sieć. Po podgrzaniu materiał przechodzi w formę sześcienną, w której pozycje środków dimerów pozostają uporządkowane na sieci, lecz same dimery nie zachowują już stałego kierunku. Zamiast tego obracają się między kilkoma preferowanymi orientacjami, spędzając czas w płytkich dolinach energii odpowiadających określonym ustawieniom. Materiał pozostaje ciałem stałym, lecz jego wewnętrzna struktura staje się dynamicznie nieuporządkowana.
Śledzenie przemiany wzdłuż gładkiej ścieżki
Aby ustalić, która faza jest bardziej stabilna w danej temperaturze, trzeba porównać ich energie swobodne, łączące energię i entropię (miarę nieuporządkowania). Standardowe metody oparte na małych drganiach wokół ustalonych pozycji zawodzą, gdy atomy przemieszczają się lub obracają znacząco. Autorzy stosują tu technikę zwaną integracją termodynamiczną naprężenie–odkształcenie, opartą na obliczeniach pierwszych zasad i dynamice molekularnej. Konstruują gładką ścieżkę deformacji, która ciągłe przekształca komórkę symulacyjną od struktury ortorombicznej do sześciennej. Wzdłuż tej ścieżki prowadzą długie symulacje w stałych temperaturach i mierzą, jak wewnętrzne naprężenie reaguje na narzucone odkształcenie. Zintegrowanie tej odpowiedzi naprężenia daje różnicę energii swobodnej między obiema fazami.
Widziane entropią poprzez ruch atomów
Obliczenia wykazują, że w około 600 K faza ortorombiczna wciąż ma niewielką przewagę, natomiast przy 650 K wygrywa faza sześcienna przewagą kilku tysięcznych elektrona wolt na jednostkę wzoru. Interpolacja tych wyników daje temperaturę przejścia około 611 K. To niżej niż szacunki eksperymentalne, ale nadal w rozsądnym porozumieniu, zważywszy na małe różnice energii swobodnej. Energia wewnętrzna fazy sześciennej jest faktycznie wyższa; to duży przyrost entropii ją stabilizuje, bezpośrednio wynikający z rotacyjnego nieuporządkowania dimerów C2. Analizując, jak orientacja każdego dimeru traci pamięć o początkowym kierunku w czasie, autorzy pokazują, że dimery zmieniają ustawienie w skali poniżej pikosekundy, zacierając rozgraniczenie między zwykłymi kategoriami entropii „drgającej” i „konfiguracyjnej”.
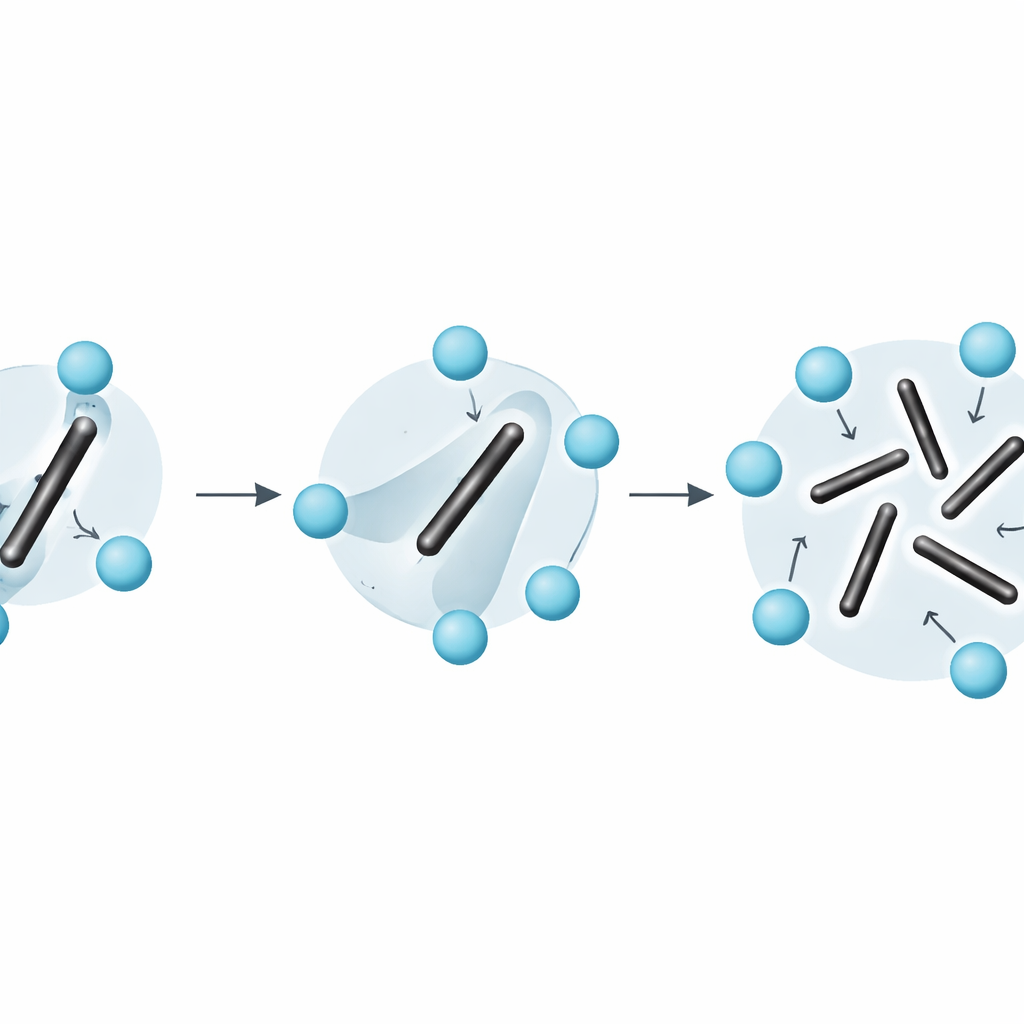
Poza prostymi obrazami nieporządku w ciałach stałych
Praca podkreśla również, że powszechne uproszczenia — takie jak traktowanie entropii jako sumy drgań wokół stałych konfiguracji plus osobne zliczanie statycznych orientacji — zawodzą w przypadku materiałów takich jak Li2C2. Ponieważ obroty dimerów są szybkie i silnie sprzężone z zwykłymi drganiami, układu nie da się czysto rozdzielić na oddzielne części „drgającą” i „przeorganizowującą się”. Metoda integracji naprężenie–odkształcenie omija tę trudność: wydobywa pełną energię swobodną bezpośrednio z mikroskopowej dynamiki, bez potrzeby zgadywania, jak entropia powinna być podzielona.
Czego uczy nas to badanie
Mówiąc wprost, badanie pokazuje, jak ciało stałe może pozostać sztywne, podczas gdy jego wewnętrzne „cegiełki” zyskują coraz większą swobodę skręcania i obracania się, oraz jak ta wewnętrzna swoboda może sprawić, że bardziej nieuporządkowana struktura stanie się termodynamicznie preferowana. Dla Li2C2 faza sześcienna o wysokiej temperaturze jest stabilizowana nie dlatego, że jest energetycznie tańsza, lecz dlatego, że oferuje znacznie więcej sposobów orientacji i ruchu dimerom C2. Udowadniając, że integracja termodynamiczna naprężenie–odkształcenie potrafi uchwycić tę subtelną równowagę między porządkiem, energią i entropią, praca otwiera drogę do przewidywania podobnych przemian w innych dynamicznie nieuporządkowanych ciałach stałych, które mogą stanowić podstawę przyszłych urządzeń chłodzących, baterii i materiałów inteligentnych.
Cytowanie: Klarbring, J., Filippov, S., Häussermann, U. et al. Ab initio determination of phase stabilities of dynamically disordered solids: rotational C2 disorder in Li2C2. Sci Rep 16, 8965 (2026). https://doi.org/10.1038/s41598-026-43795-z
Słowa kluczowe: przejście fazowe w stanie stałym, dynamiczne nieuporządkowanie, dynamika molekularna, węgliki litu, integracja termodynamiczna