Clear Sky Science · pl
Dane kierujące projektowaniem blokerów LNA do skutecznego usuwania zanieczyszczeń w bibliotekach Ribo‑Seq
Dlaczego oczyszczanie danych sekwencyjnych ma znaczenie
Nowoczesna biologia często polega na odczytywaniu milionów drobnych fragmentów RNA, aby zrozumieć, jak komórki wytwarzają białka. Jednak te potężne pomiary, w szczególności metoda zwana profilowaniem rybosomów (Ribo‑Seq), mogą być zaśmiecone przez nieistotne fragmenty RNA, które marnują moc sekwencjonowania i pieniądze. W tym badaniu opisano prostą, opartą na danych metodę projektowania wyspecjalizowanych molekularnych „blokerów”, które selektywnie usuwają niechciane fragmenty, niemal podwajając użyteczną informację uzyskiwaną z tego samego eksperymentu.
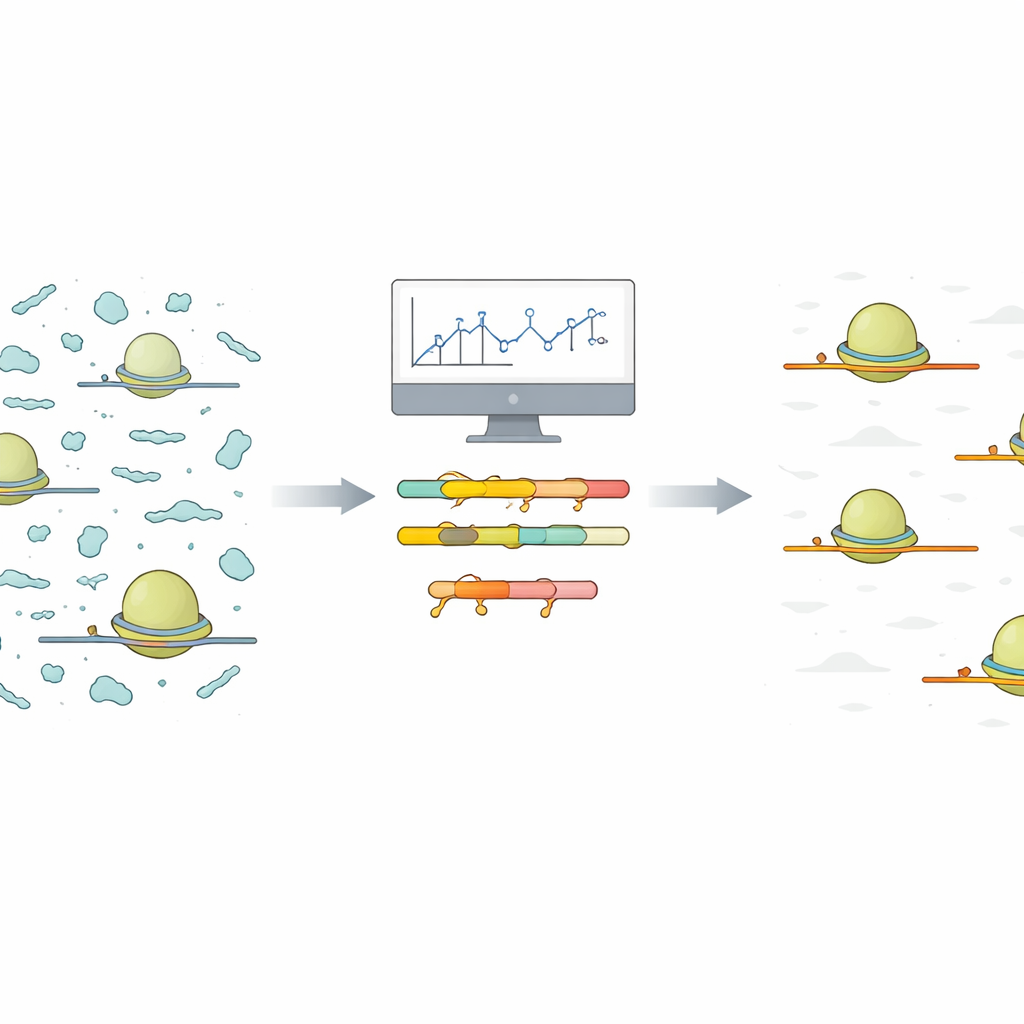
Problem hałaśliwych migawków rybosomów
Ribo‑Seq rejestruje chwilowy obraz tego, które komunikaty w komórce są aktywnie tłumaczone na białka. Aby to zrobić, naukowcy izolują rybosomy wraz z krótkimi odcinkami mRNA, które chronią. Wszystko inne jest rozkładane, a chronione fragmenty są sekwencjonowane i mapowane z powrotem do genomu. W praktyce jednak wiele innych małych fragmentów ncRNA (niekodującego RNA) przenika przez ten proces. Ponieważ te zanieczyszczenia są liczne i wysoce zmienne, pochłaniają dużą część odczytów sekwencyjnych, pozostawiając mniej odczytów na prawdziwe sygnały kodujące białka, którymi interesują się badacze.
Dlaczego istniejące triki oczyszczania zawodzą
Standardowe strategie próbują usunąć obfite rybosomalne i inne ncRNA za pomocą wcześniej zaprojektowanych sond wychwytujących lub enzymów. Metody te działają dobrze, gdy docelowe RNA są nienaruszone i przewidywalne, ale Ribo‑Seq celowo rozcina RNA na wiele fragmentów o różnej długości. To fragmentowanie miesza miejsca docelowe dla zestawów stałych sond, co znacznie obniża efektywność deplecji. Dodatkowo dokładny zestaw zanieczyszczeń zależy od badanej gatunku, warunków wzrostu, a nawet użytego enzymu nukleolitycznego. Istniejące procedury oczyszczania zwykle wymagają też wielu inkubacji i kroków oczyszczania, co zabiera czas i może powodować utratę próbki lub wprowadzać zniekształcenia.
Blokery niestandardowe zaprojektowane na podstawie rzeczywistych danych
Autorzy proponują uproszczone podejście, które zaczyna się od krótkiego, niskokosztowego próbnego sekwencjonowania przeprowadzonego w tych samych warunkach, które planuje się zastosować w pełnym eksperymencie. Udostępniają skrypt w R, który bierze wyrównane odczyty z tego pilota i automatycznie grupuje podobne fragmenty zanieczyszczeń na podstawie sekwencji. Dla każdej grupy skrypt raportuje najkrótszą wspólną sekwencję występującą w fragmentach. Krótkie, wspólne odcinki są idealnymi miejscami docelowymi dla wyspecjalizowanych cząsteczek zwanych oligonukleotydami z zamkniętymi nukleotydami (LNA). LNA to krótkie nici z modyfikacją chemiczną, która powoduje silne wiązanie z odpowiadającym RNA. Skrypt generuje też czytelne mapy cieplne i wykresy podsumowujące, pomagając użytkownikom zobaczyć, które zanieczyszczenia dominują i ile celów LNA byłoby potrzebnych do znaczącego oczyszczenia.
Jednostopniowe oczyszczanie podczas amplifikacji
Zamiast fizycznie usuwać zanieczyszczenia z próbki, metoda używa oligonukleotydów LNA jako blokerów podczas etapu amplifikacji DNA, który buduje bibliotekę sekwencyjną. Autorzy testowali dodawanie tych blokerów albo podczas początkowej odwrotnej transkrypcji, albo podczas późniejszej amplifikacji PCR. Stwierdzili, że dodanie LNA podczas amplifikacji było bardziej efektywne i wymagało niższych stężeń, redukując testowy zanieczyszczający fragment ponad tysiąckrotnie, działając niezależnie od orientacji nici. Praktyczne wskazówki projektowe obejmują naprzemienne stosowanie standardowych bloków DNA i LNA, użycie długości minimalnej 14 jednostek dla rośliny Arabidopsis oraz modyfikację końca ogonowego, aby sam bloker nie mógł być przypadkowo przedłużony.
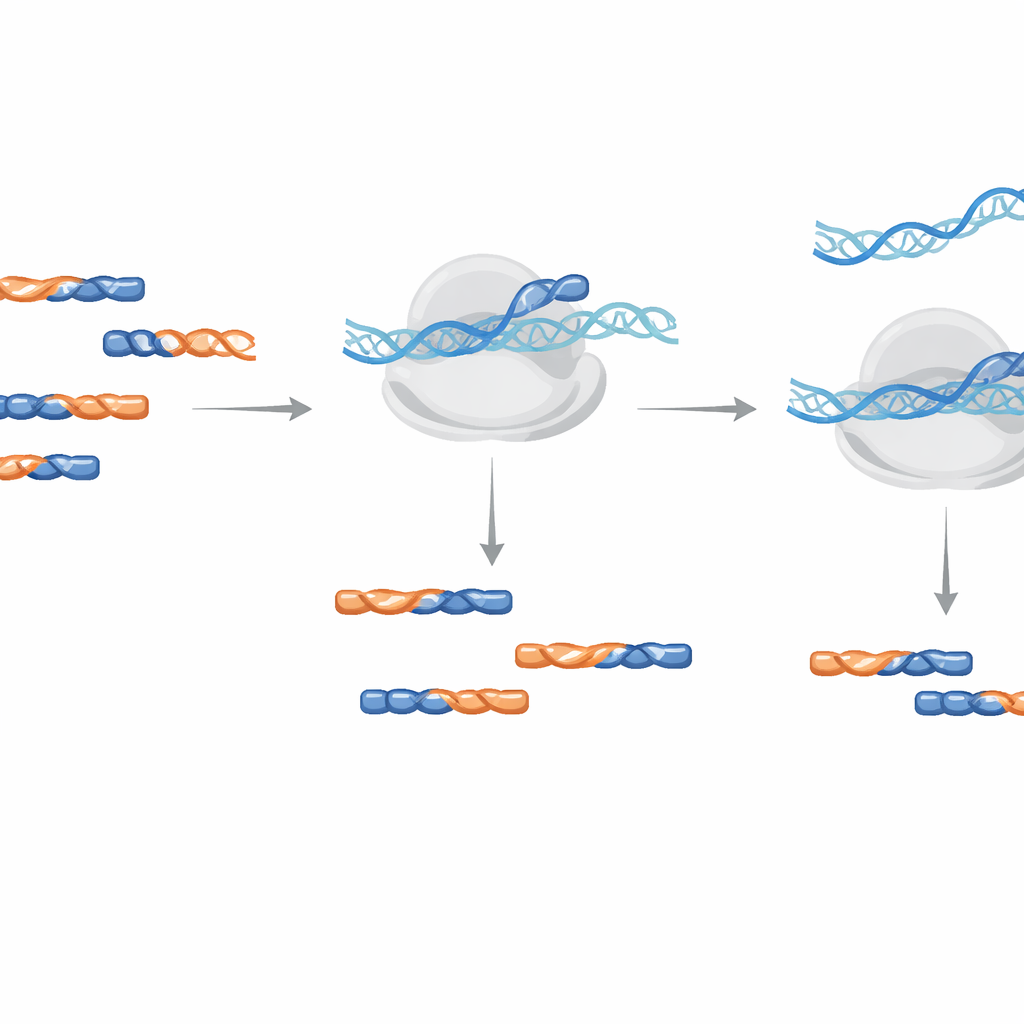
Więcej użytecznych odczytów bez zniekształcania sygnału
Aby pokazać wydajność w praktyce, zespół zaprojektował pięć blokerów LNA skierowanych przeciwko najczęstszym grupom zanieczyszczeń obserwowanych w typowych warunkach wzrostu u roślin Arabidopsis. Po dodaniu tej mieszanki podczas amplifikacji bibliotek, odsetek zidentyfikowanych zanieczyszczeń spadł o ponad 30%, a liczba użytecznych odczytów kodujących białka niemal się podwoiła. Co ważne, porównanie liczby odczytów na poziomie genu między bibliotekami z i bez traktowania LNA wykazało niemal idealną zgodność, co wskazuje, że blokery usunęły fragmenty śmieciowe bez zniekształcania biologicznego sygnału pochodzącego z autentycznych odcisków mRNA.
Co to oznacza dla przyszłych eksperymentów
Ta praca pokazuje, że krótki eksperyment pilotażowy, w połączeniu z łatwym w użyciu skryptem analitycznym i niewielkim zestawem dopasowanych blokerów LNA, może przekształcić zaśmiecone biblioteki Ribo‑Seq w znacznie czystsze, bardziej informatywne zbiory danych w jednym kroku pipetowania. Badacze zyskują więcej znaczących odczytów na serię, oszczędzając koszty i upraszczając projekt eksperymentu, przy jednoczesnym zachowaniu dokładnych pomiarów translacji genów. Autorzy udostępniają też gotowe profile zanieczyszczeń i projekty blokerów dla typowych warunków roślinnych oraz sugerują, że podobne zasoby można opracować dla wielu organizmów, co uczyni wysokiej jakości profilowanie rybosomów bardziej dostępnym w całej społeczności badawczej.
Cytowanie: Ricciardi, D.A., Peter, F.E. & Böhmer, M. Data-driven design of LNA-blockers for efficient contaminant removal in Ribo-Seq libraries. Sci Rep 16, 8565 (2026). https://doi.org/10.1038/s41598-026-43117-3
Słowa kluczowe: profilowanie rybosomów, zanieczyszczenia RNA, zamknięte kwasy nukleinowe (LNA), oczyszczanie bibliotek sekwencyjnych, regulacja translacji