Clear Sky Science · pl
Odkrycie naturalnych analogów apigeniny jako inhibitorów demetylazy specyficznej dla lizyny 1 przeciw nowotworowym komórkom zarodkowym jądra
Dlaczego te badania mają znaczenie dla zdrowia mężczyzn
Nowotwory zarodkowe jądra są najczęstszymi nowotworami jąder u młodych mężczyzn; choć wielu pacjentów można wyleczyć, obecne terapie często zagrażają płodności i powodują silne skutki uboczne. W tym badaniu sprawdzono, czy naturalny związek spokrewniony z barwnikiem roślinnym apigeniną, występującym w owocach i warzywach, można przekształcić w precyzyjny lek hamujący wzrost komórek nowotworowych jądra przy jednoczesnym oszczędzaniu zdrowej tkanki jądra.
Naturalny pomysł z codziennych roślin
Apigenina to żółty barwnik roślinny obecny w takich produktach jak pietruszka, seler czy rumianek. Chemicy od dawna wiedzą, że może wpływać na zachowanie komórek, lecz jej przydatność wobec nowotworów zarodkowych jądra nie była dotąd zbadana. Autorzy zaczęli od złożenia niewielkiej biblioteki naturalnych związków podobnych do apigeniny. Skoncentrowali się na białku zwanym LSD1 — enzymie pomagającym kontrolować, które geny są włączane lub wyłączane poprzez subtelne modyfikacje białek pakujących DNA. Ponieważ poziomy LSD1 są nieprawidłowo wysokie w kilku nowotworach, w tym w nowotworach zarodkowych jądra, blokowanie tego enzymu wydaje się atrakcyjną strategią spowalniającą rozwój guza.
Poszukiwanie najskuteczniejszego blokeru pochodzenia roślinnego
Naukowcy systematycznie przetestowali szesnaście naturalnych analogów apigeniny, aby sprawdzić, jak silnie każdy z nich potrafi zahamować LSD1 w teście in vitro. Wiele związków wykazało pewne działanie, ale jeden wyróżniał się szczególnie: wariant nazwany 8,3’-diprenylapigeniną był najsilniejszym inhibitorem, działając w niskich stężeniach mikromolowych. Porównując cechy chemiczne wszystkich analogów, zespół odtworzył, które drobne modyfikacje strukturalne wzmacniają lub osłabiają aktywność hamującą enzym. Stwierdzili, że pewna grupa chemiczna, znana jako grupa 7-hydroksylowa, jest szczególnie ważna dla przyczepności do LSD1, a dodanie masywnych łańcuchów „prenylowych” w określonych pozycjach znacząco zwiększało moc działania.
Jak nowy związek atakuje komórki nowotworowe
Następnie zespół przeszedł od testów enzymatycznych do badań na żywych komórkach. Ekspozycja ludzkich linii komórkowych nowotworów zarodkowych jądra, NCCIT i NTERA-2, na 8,3’-diprenylapigeninę znacząco zmniejszyła zdolność tych komórek do mnożenia się, również w relatywnie niskich stężeniach. Dla porównania, te same dawki zastosowane wobec dwóch normalnych linii komórkowych jądra, TM3 i TM4, praktycznie ich nie uszkodziły. Ta selektywność sugeruje, że komórki nowotworowe, które silnie polegają na nadaktywności LSD1, są znacznie bardziej wrażliwe na jego hamowanie niż komórki normalne — co jest obiecującą wskazówką dla potencjalnych przyszłych terapii.
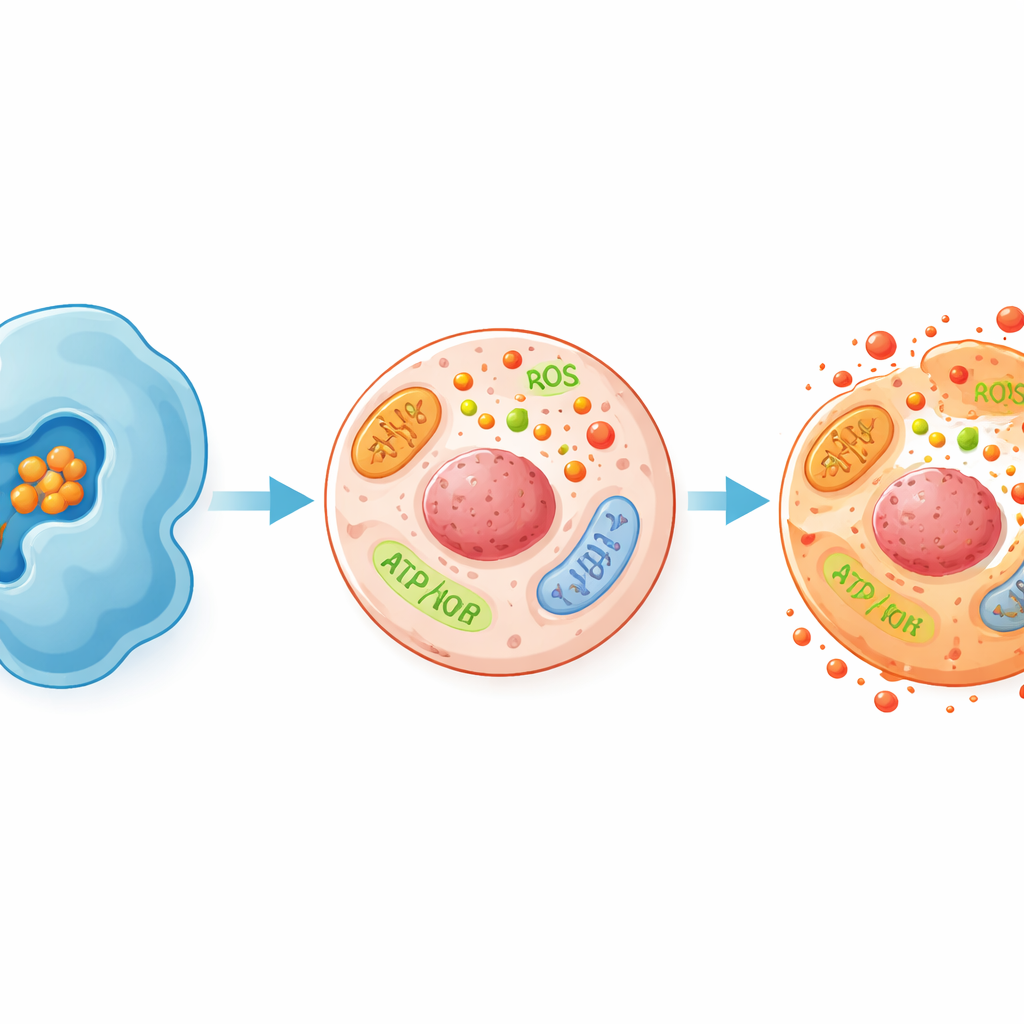
Zaglądając do odpowiedzi stresowej komórki
Aby zrozumieć, co dzieje się wewnątrz komórek nowotworowych, gdy LSD1 jest zablokowany przez 8,3’-diprenylapigeninę, naukowcy zmierzyli kilka markerów stresu komórkowego. Stwierdzili, że leczone komórki NTERA-2 wytwarzały więcej reaktywnych form tlenu — wysoko reaktywnych cząsteczek mogących uszkadzać białka, lipidy i DNA. Jednocześnie aktywność katalazy, enzymu ochronnego rozkładającego szkodliwe utleniacze, zmniejszyła się. Poziom ATP, „waluty” energetycznej komórki, również spadł, co wskazuje na zaburzenie produkcji energii. Inne wskaźniki uszkodzeń — w tym przeciekanie enzymu LDH z komórek, zwiększona aktywność enzymu antyoksydacyjnego SOD oraz wyższe poziomy markera uszkodzeń lipidów MDA — rosły w sposób zależny od dawki i czasu ekspozycji. Razem te zmiany pokazują obraz komórek nowotworowych popchniętych w stres oksydacyjny, z którym nie potrafią sobie poradzić, co ostatecznie prowadzi do ich śmierci.
Od dopasowania molekularnego do możliwego leku
Symulacje komputerowe pomogły wyjaśnić, dlaczego 8,3’-diprenylapigenina działa tak skutecznie. Badania dokowania i dynamiki pokazały związek ciasno osadzony w kieszeni LSD1, tworzący stabilizujące wiązania wodorowe i kontakty hydrofobowe, przy czym wiązanie jest odwracalne. Ta odwracalność jest ważna, ponieważ niektóre wcześniejsze, nieodwracalne inhibitory LSD1 powodowały działania niepożądane dotyczące krwi i układu odpornościowego w badaniach klinicznych. W tym przypadku 8,3’-diprenylapigenina silnie i selektywnie hamowała LSD1, nie wpływając znacząco na blisko spokrewnione enzymy MAO-A i MAO-B, co zmniejsza ryzyko niepożądanych działań w mózgu i innych tkankach.
Co to może znaczyć dla pacjentów
Mówiąc prosto, praca ta identyfikuje cząsteczkę pochodzenia roślinnego, która potrafi precyzyjnie trafić w enzym związany z rakiem w komórkach nowotworowych jądra, zahamować ich wzrost i wywołać kontrolowaną formę samozniszczenia, przy jednoczesnym pozostawieniu normalnych komórek jądra w dużej mierze nietkniętych w badaniach laboratoryjnych. Choć potrzebne są dalsze badania — w tym badania na zwierzętach i testy bezpieczeństwa u ludzi — 8,3’-diprenylapigenina stanowi obiecujący punkt wyjścia („szkielet”) do projektowania łagodniejszych, bardziej ukierunkowanych terapii nowotworów zarodkowych jądra, które mogą lepiej chronić płodność i ogólne zdrowie.
Cytowanie: Sun, LW., Zhang, M., Li, CF. et al. Discovery of natural apigenin analogues as lysine-specific demethylase 1 inhibitors against tumoral testicular germ cells. Sci Rep 16, 8917 (2026). https://doi.org/10.1038/s41598-026-42263-y
Słowa kluczowe: rak jądra, inhibitory LSD1, apigenina, stres oksydacyjny, terapia celowana