Clear Sky Science · pl
Hybrydowy, dostrojony model głębokiego uczenia do diagnozy raka piersi z wykorzystaniem danych genetycznych
Dlaczego to ma znaczenie dla pacjentów i rodzin
Rak piersi jest obecnie najczęściej rozpoznawanym nowotworem u kobiet na całym świecie, a wczesne wykrycie może przesądzać o życiu lub śmierci. Lekarze coraz częściej mają dostęp do informacji genetycznej pacjenta, jednak przekształcenie dziesiątek tysięcy pomiarów genów w jasne odpowiedzi jest niezwykle trudne. Artykuł opisuje nowy model komputerowy, który czyta te złożone wzory genetyczne, aby wykryć raka piersi i przewidywać przebieg choroby z imponującą dokładnością, dając klinicystom potencjalnie potężne narzędzie do wcześniejszych i bardziej wiarygodnych decyzji.
Od genów do sygnałów ostrzegawczych
Każdy guz piersi ma molekularny odcisk palca zakodowany w aktywności tysięcy genów. Autorzy postawili sobie za cel zbudowanie systemu potrafiącego czytać ten odcisk bezpośrednio, zamiast polegać tylko na obrazach czy garstce dobrze znanych genów, takich jak BRCA1 i BRCA2. Pracowali z dwoma z największych publicznych zasobów w genomice nowotworów: kohortą raka piersi TCGA, która zawiera aktywność 17 814 genów w 590 próbkach, oraz badaniem METABRIC, obejmującym dane genomowe i kliniczne dla ponad 1 400 pacjentów. Ich cel był ambitny: zaprojektować metodę zdolną poradzić sobie z tym napływem informacji, znaleźć najbardziej wymowne sygnały i nadal działać niezawodnie w całkowicie odrębnych grupach pacjentów.
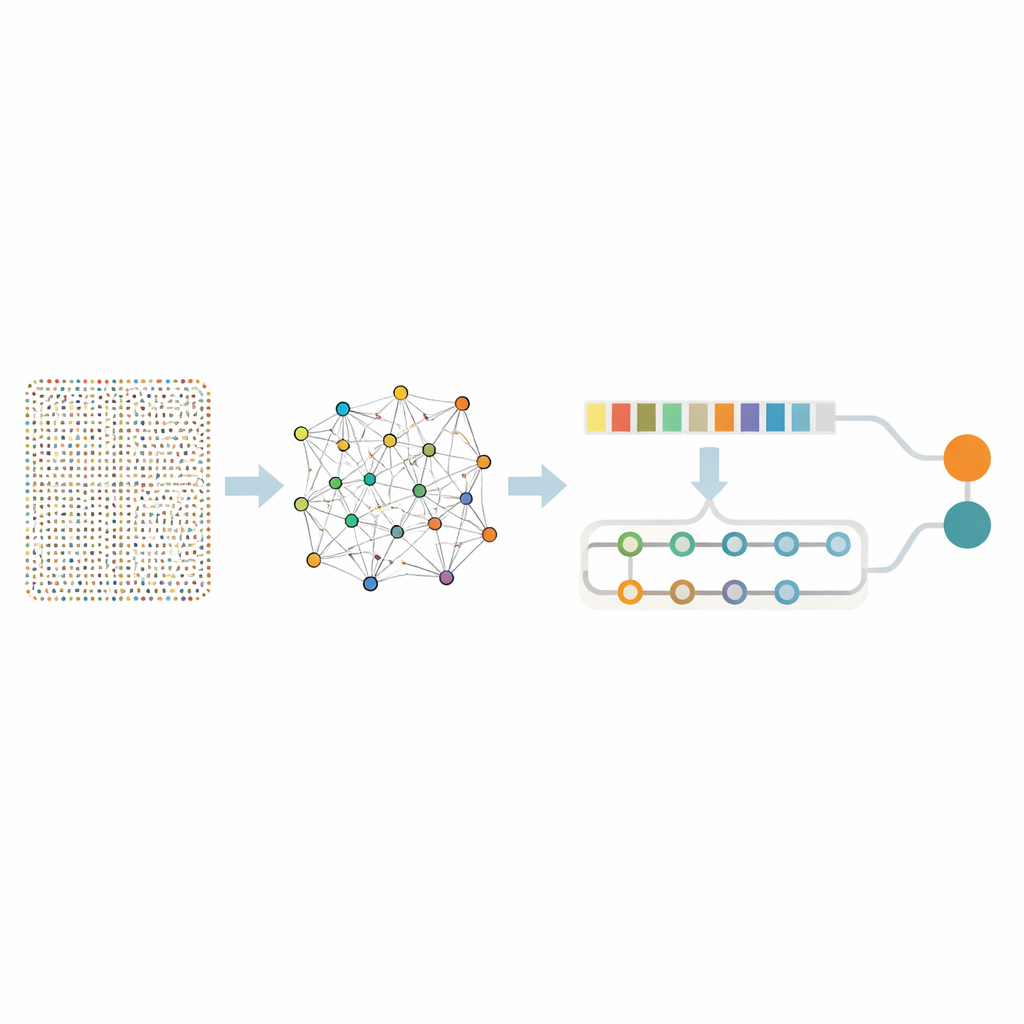
Redukcja tysięcy genów do użytecznego zestawu
Analiza niemal osiemnastu tysięcy genów naraz jest przytłaczająca nawet dla zaawansowanych algorytmów i grozi wychwyceniem bezsensownego szumu. Badacze zastosowali więc dwuetapowe „sito”, aby wyizolować mniejszy zestaw rzeczywiście informatywnych genów. Najpierw użyli techniki zwanej Random Forest, która pyta wiele drzew decyzyjnych, które geny są najważniejsze dla rozróżnienia tkanki nowotworowej od zdrowej. Ten krok zredukował listę do 436 obiecujących genów. Następnie przeanalizowali, jak te geny zachowują się razem, stosując wydobywanie reguł asocjacyjnych — metodę wykrywającą grupy genów, które mają tendencję do aktywacji jednocześnie w guzach. Ta dodatkowa warstwa analizy zidentyfikowała pary genów i sieci powiązane z kluczowymi procesami nowotworowymi, takimi jak szybka podział komórek, naprawa uszkodzeń DNA oraz zmiany w otaczającej guza tkance. Po tym zawężeniu pozostało 332 geny — nadal bogate w znaczenie biologiczne, ale znacznie bardziej przystępne do głębszej analizy.
Dwuczęściowa sieć neuronowa ucząca się wzorców i kontekstu
Mając w ręku wyselekcjonowany zestaw genów, zespół zbudował hybrydowy model głębokiego uczenia łączący dwa typy sieci neuronowych. Jedna część, znana jako sieć konwolucyjna, skanuje listę genów, aby wychwycić lokalne wzorce — klastry genów, które mają tendencję do jednoczesnego wzrostu lub spadku. Druga część, dwukierunkowa sieć pamięciowa, analizuje te same dane, śledząc relacje na długim dystansie i rejestrując, jak odległe geny wpływają na siebie w całym profilu. Przed treningiem autorzy zrównoważyli dane tak, by próbki nowotworowe i nienowotworowe były reprezentowane uczciwie, oraz dodali niewielkie ilości sztucznego szumu, ucząc model, by nie dał się zwieść losowym fluktuacjom.
Jak system sprawdza się w testach z rzeczywistych danych
Po treningu i testach na danych TCGA hybrydowa sieć poprawnie odróżniała próbki guzowe od prawidłowych z około 97% dokładnością i niemal doskonałą zdolnością separacji obu grup. Co ważne, przewyższała prostsze układy głębokiego uczenia oraz standardowe narzędzia uczenia maszynowego, takie jak regresja logistyczna i maszyny wektorów nośnych, nawet gdy te konkurencyjne metody otrzymały ten sam starannie dobrany zestaw genów. Najważniejszym sprawdzianem była jednak odporność modelu na zupełnie inny zbiór danych. Zastosowany do METABRIC, zbieranego w innych szpitalach przy użyciu innych metod laboratoryjnych, system utrzymał wysoką skuteczność: w najlepszym przebiegu osiągnął 99,3% dokładności i prawidłowo zidentyfikował wszystkich pacjentów, którzy później zmarli z powodu raka piersi — kluczowa cecha, jeśli narzędzie ma służyć do wyłapywania przypadków wysokiego ryzyka.
Co to może znaczyć dla przyszłej opieki
Dla osoby niebędącej specjalistą sedno sprawy jest takie: badanie dostarcza inteligentnego filtra i czytnika danych genetycznych, który z niezwykłą konsekwencją może wykrywać raka piersi i związane z nim ryzyko w dużych grupach pacjentów. Łącząc przemyślaną strategię selekcji genów z dwugalążkową siecią neuronową, autorzy pokazują, że komputery potrafią wydobywać klinicznie istotne sygnały z ogromnych zestawów genetycznych, nie tylko w jednym badaniu, ale także pomiędzy niezależnymi kohortami. Choć konieczne są dalsze badania, by przetestować podejście w różnorodnych populacjach i wyjaśnić jego decyzje w szczegółach, metoda wskazuje drogę ku przyszłości, w której prosty pobrany z krwi lub tkanki materiał mógłby zasilać takie modele i pomagać lekarzom we wcześniejszym wykrywaniu guzów oraz precyzyjniejszym doborze terapii.
Cytowanie: Hesham, F., Abbassy, M.M. & Abdalla, M. Hybrid tuned deep learning model for breast cancer diagnosis using genetic data. Sci Rep 16, 9664 (2026). https://doi.org/10.1038/s41598-026-41643-8
Słowa kluczowe: genomika raka piersi, diagnoza przy użyciu głębokiego uczenia, biomarkery ekspresji genów, wczesne wykrywanie nowotworów, wsparcie decyzji klinicznych