Clear Sky Science · pl
DPAS: wskaźnik anomalii peptydów związanych z chorobą do identyfikacji peptydów patogennych za pomocą uczenia jednoklasowego
Dlaczego małe fragmenty białek mają znaczenie dla naszego zdrowia
Peptydy — krótkie odcinki białek — stały się gwiazdami współczesnej medycyny. Mogą działać jako precyzyjne nośniki informacji w organizmie i coraz częściej wykorzystywane są jako leki oraz markery chorób. Jednak ustalenie, które peptydy rzeczywiście wiążą się z chorobą, zwykle wymaga jasnych przykładów zarówno „chorobowych”, jak i „niechorobowych” peptydów, czego biologii rzadko udaje się dostarczyć. W tym badaniu przedstawiono nowe podejście do wykrywania potencjalnie szkodliwych peptydów, wykorzystujące wyłącznie te, które już znamy jako związane z chorobą, co daje szybszą i mniej obarczoną uprzedzeniami drogę do odkrywania przyszłych diagnostyk i terapii.
Problem znalezienia grupy „niechorobowej”
Tradycyjne modele komputerowe uczą się poprzez porównanie dwóch stron: przykładów pozytywnych, znanych jako związane z chorobą, oraz przykładów negatywnych, które uważa się za nieszkodliwe. W badaniach nad peptydami ta druga grupa jest problematyczna. Wiele peptydów po prostu nie zostało przetestowanych, więc nadawanie im etykiety „niechorobowy” może być mylące i wprowadzać uprzedzenia. Poprzednie prace nad peptydami przeciwnowotworowymi czy przeciwzapalnymi osiągały imponującą dokładność, ale często opierały się na ręcznie zbudowanych lub domniemanych zbiorach negatywnych. W efekcie ich modele mogą mieć trudności z wykrywaniem rzadkich sygnałów lub nowych rodzajów peptydów chorobotwórczych, które nie przypominają danych treningowych.
Uczenie się na tym, co wiemy, zamiast na tym, co zgadujemy
Autorzy podążają inną drogą: zamiast wymuszać problem dwustronny, traktują peptydy związane z chorobą jako jedną spójną grupę i pytają: „Jak ta grupa wygląda szczegółowo?” Zgromadzili ponad 760 000 zmutowanych ludzkich peptydów z wyspecjalizowanej bazy danych związanej z rakiem i opisali każdy peptyd za pomocą bogatego zestawu cech. Należą do nich częstość występowania poszczególnych aminokwasów, układy par aminokwasów, podstawowe cechy fizykochemiczne takie jak objętość czy hydrofilowość oraz krótkie powtarzające się wzorce sekwencyjne zwane motywami. Technika zwana analizą głównych składowych (PCA) następnie kompresuje ten wysokowymiarowy opis do bardziej zwięzłej postaci, zachowując główne źródła zmienności.
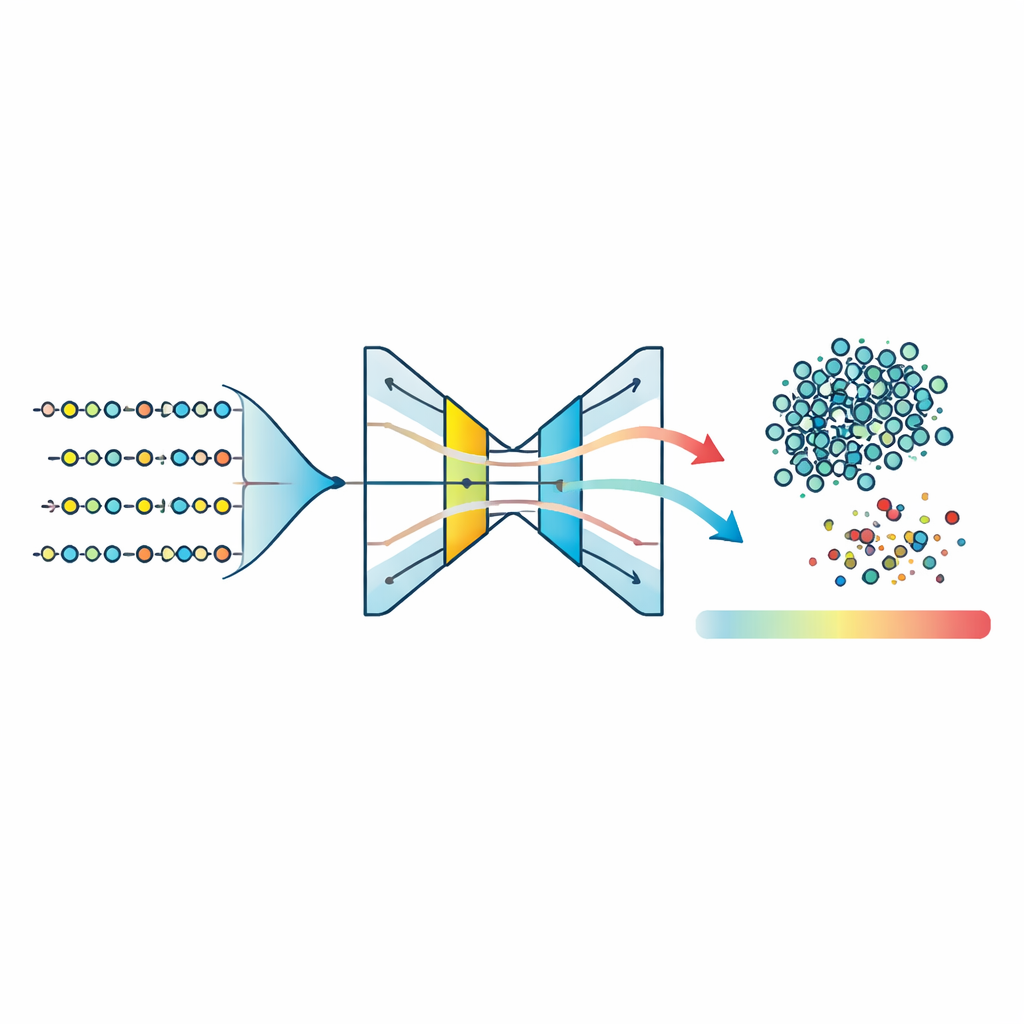
Wykrywanie nietypowych peptydów za pomocą modeli jednoklasowych
W tak skompresowanej przestrzeni cech zespół wytrenował trzy modele „jednoklasowe” — algorytmy zaprojektowane do nauki kształtu pojedynczej grupy i oznaczania wszystkiego, co się nie mieści. Przetestowali One-Class Support Vector Machines, Isolation Forests oraz rodzaj sieci neuronowej zwany autoenkoderem. Autoenkoder uczy się ściskać cechy każdego peptydu do wąskiej wewnętrznej reprezentacji, a następnie je rekonstruować; peptydy należące do wyuczonego wzorca chorobowego są odtwarzane dokładnie, natomiast nietypowe generują wyższy błąd rekonstrukcji. Porównanie znormalizowanych wskaźników anomalii we wszystkich metodach pokazuje, że autoenkoder tworzy najciaśniejszy klaster typowych peptydów i naj wyraźniejsze rozdzielenie między elementami zgodnymi a odchyleniami. Ustalając próg błędu rekonstrukcji wokół 95. percentyla, model klasyfikuje większość peptydów jako prawdopodobnie związane z chorobą, jednocześnie konsekwentnie wskazując niewielki ułamek jako nietypowe.
Przekształcenie złożonych wskaźników w jedną, znaczącą liczbę
Aby ułatwić interpretację biologiczną wyników, autorzy wprowadzają Disease Peptide Anomaly Score (DPAS). Ten wskaźnik łączy dwa składniki: jak nietypowo peptyd wygląda dla autoenkodera (jego znormalizowany błąd rekonstrukcji) oraz jak silnie jego cechy wpływają na przewidywania, mierzone popularną metodą wyjaśniającą o nazwie SHAP. W praktyce jako szczególnie informatywne ujawniają się motywy i określone cechy fizykochemiczne. DPAS łączy te sygnały tak, że peptydy zarówno strukturalnie odmienione, jak i poparte biologicznie istotnymi cechami otrzymują wyższe pozycje. Najwyżej punktowane peptydy są następnie analizowane narzędziem wyszukującym motywy, które łączy je z znanymi sygnaturami funkcjonalnymi, takimi jak miejsca fosforylacji, regiony wiążące metale i inne wzorce regulatorowe powszechnie zaangażowane w sygnalizację i kontrolę enzymatyczną.
Co to oznacza dla przyszłych diagnostyk i leków
Mówiąc prościej, ta praca oferuje sprytniejszy filtr do znajdowania podejrzanych peptydów bez udawania, że znamy te, które na pewno są nieszkodliwe. Ucząc się wyłącznie na potwierdzonych przykładach związanych z chorobą, a następnie oceniając nowe kandydatury za pomocą DPAS, badacze mogą priorytetyzować krótką, biologicznie wiarygodną listę peptydów do badań laboratoryjnych. Wiele z najwyżej ocenionych kandydatur zawiera dobrze znane motywy funkcjonalne, co wzmacnia przypuszczenie, że mogą odgrywać role w procesach chorobowych. Choć metoda nadal opiera się na założeniach i brakuje jej do pełnej walidacji eksperymentalnie potwierdzonych „bezpiecznych” peptydów, dostarcza bardziej realistycznych i przejrzystych podstaw dla odkrywania biomarkerów peptydowych i może zostać dostosowana do innych typów danych biologicznych, w których rzetelne przykłady negatywne są rzadkie.
Cytowanie: Khalid, Z., Khalid, R. & Sezerman, O.U. DPAS: disease-associated peptide anomaly score for identifying pathogenic peptides via one-class learning. Sci Rep 16, 9170 (2026). https://doi.org/10.1038/s41598-026-40099-0
Słowa kluczowe: peptydy związane z chorobą, wykrywanie anomalii, autoenkoder, odkrywanie biomarkerów, uczenie jednoklasowe