Clear Sky Science · pl
Interpretowalny uczenie maszynowe wyjaśnia hamowanie anhydrazy węglanowej przez predykcję conformalną i kontrfaktyczną
Dlaczego bardziej inteligentne leki przeciwnowotworowe mają znaczenie
Leki przeciwnowotworowe często zachowują się jak tępe narzędzia: choć atakują komórki nowotworowe, mogą też uszkadzać zdrowe tkanki i powodować poważne skutki uboczne. Jednym z obiecujących sposobów wyostrzenia tego działania jest blokowanie określonych wariantów enzymu zwanego anhydrazą węglanową, który pomaga guzom przetrwać w warunkach niskiego poziomu tlenu. Jednak kilka wariantów tego enzymu wygląda niemal identycznie, co utrudnia projektowanie leków uderzających w „złe” izoformy w guzie bez naruszania „dobrej” izoformy występującej powszechnie w organizmie. W badaniu pokazano, jak interpretowalne metody uczenia maszynowego mogą pomóc naukowcom poruszać się po tym wyzwaniu i projektować bardziej selektywne, bezpieczniejsze kandydaty na leki.
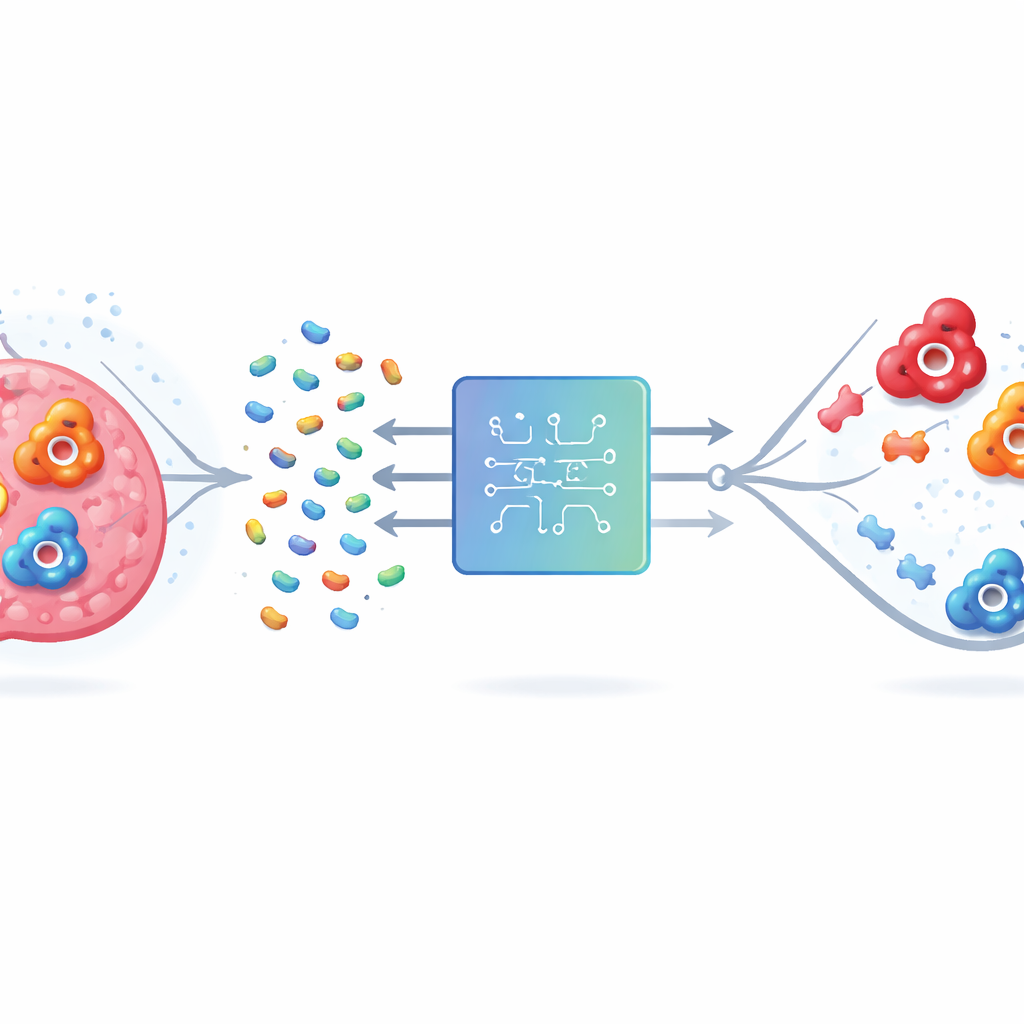
Problem trafiania w niewłaściwy cel
Ludzka anhydraza węglanowa (hCA) występuje w wielu formach, czyli izoformach. Dwie z nich, IX i XII, wiążą się z przeżywalnością komórek nowotworowych w guzach pozbawionych tlenu, więc ich blokada mogłaby spowalniać chorobę i poprawiać leczenie. Jednak izoforma II jest powszechna w zdrowych tkankach i ma centrum aktywne wyglądające bardzo podobnie do IX i XII. Leki wiążące wszystkie trzy mogą wywołać niepożądane problemy, takie jak kwasica metaboliczna czy zaburzenia widzenia. Tradycyjne metody laboratoryjne i komputerowe mają trudności, ponieważ enzymy to duże, złożone molekuły, a liczba możliwych związków o właściwościach leków jest astronomicznie wielka. Wypróbowanie ich wszystkich, w laboratorium lub obliczeniowo, po prostu nie jest możliwe.
Budowanie czystej i wiarygodnej bazy danych
Autorzy rozwiązali to najpierw przez zgromadzenie starannie oczyszczonej bazy danych obejmującej tysiące cząsteczek testowanych przeciwko hCA II, IX i XII z repozytorium ChEMBL. Ustandaryzowali struktury chemiczne, usunęli wątpliwe pomiary i skupili się na związkach zawierających typową dla tej klasy inhibitorów grupę wiążącą cynk. Przy zastosowaniu rygorystycznych progów oznaczyli cząsteczki jako wyraźnie aktywne lub wyraźnie nieaktywne i odrzucili przypadki na granicy, które mogłyby wprowadzić modele w błąd. Ponieważ było znacznie więcej cząsteczek nieaktywnych niż aktywnych, zbalansowali dane, aby algorytmy uczenia nie preferowały po prostu klasy dominującej. Zastosowali też sposób dzielenia danych oparty na szkielecie („scaffold”), tak aby zestawy treningowe i testowe zawierały różne podstawowe ramy molekularne, co dało bardziej realistyczny obraz tego, jak modele poradzą sobie z naprawdę nowymi związkami.
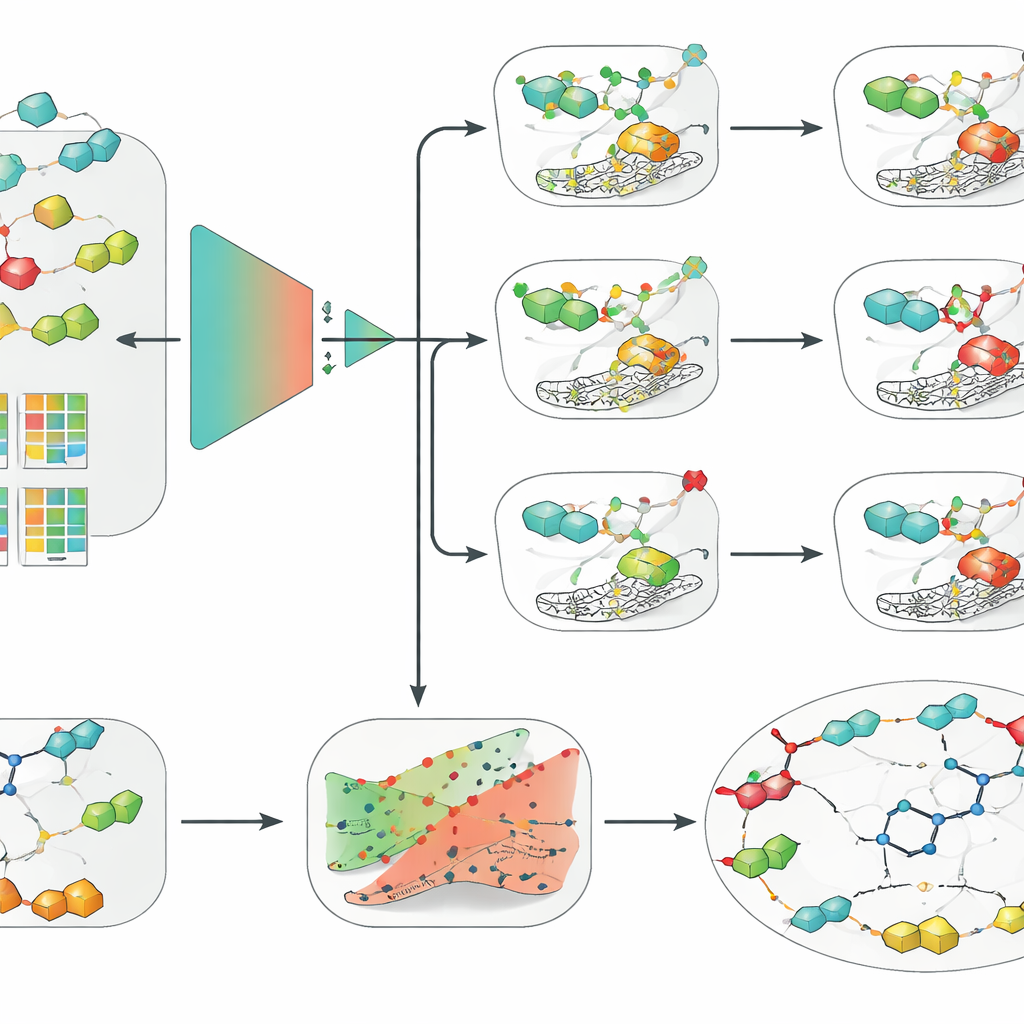
Proste modele przewyższają głębokie uczenie przy ograniczonych danych
Z tą oczyszczoną bazą danych zespół porównał szeroki zakres podejść — od klasycznych metod uczenia maszynowego, takich jak regresja logistyczna, lasy losowe i maszyny wektorów nośnych (SVM), po współczesne sieci neuronowe głębokiego uczenia, w tym modele grafowe operujące bezpośrednio na strukturach molekularnych. Połączyli je z kilkoma sposobami kodowania cząsteczek, takimi jak tradycyjne deskryptory ręcznie opracowane, odciski palców oparte na kluczach oraz wyuczone osadzenia z modelu języka chemicznego. We wszystkich trzech izoformach i przy surowszej ocenie opartej na szkielecie konsekwentnie wyróżniała się jedna kombinacja: SVM zasilony odciskami rozszerzonej łączności (extended-connectivity fingerprints), czyli uporządkowanym sposobem opisu lokalnych środowisk chemicznych w cząsteczce. Co zaskakujące, ta stosunkowo prosta konfiguracja przewyższyła bardziej modowe modele grafowe i głębokiego uczenia, podkreślając, że jakość danych, staranna walidacja i dobre deskryptory molekularne mogą mieć większe znaczenie niż złożoność algorytmiczna przy umiarkowanej wielkości zbiorów danych.
Dodanie niezawodnej pewności i przyjaznych wyjaśnień
Naukowcy następnie opakowali swój najlepszy model SVM w dwie dodatkowe warstwy zaprojektowane tak, by uczynić jego przewidywania bardziej użytecznymi w rzeczywistym odkrywaniu leków. Najpierw zastosowali ramę zwaną predykcją conformalną, która nie tylko zwraca pojedynczą odpowiedź tak/nie, lecz dostarcza region prawdopodobnych wyników wraz z gwarantowaną stopą błędu. Pozwala to naukowcom regulować, jak ostrożny ma być model, i rozpoznawać przypadki, w których model jest rzeczywiście niepewny. Po drugie, wykorzystali wyjaśnienia kontrfaktyczne, aby uczynić rozumowanie modelu bardziej intuicyjnym. Dla danej cząsteczki generowali bliskie analogi, które odwracają przewidywany wynik z aktywnego na nieaktywny lub odwrotnie. Analiza takich par dla kandydata klinicznego SLC-0111, który selektywnie blokuje IX i XII, ale nie II, niezależnie odnalazła ważny wniosek chemii medycznej: drobne zmiany w „ogonowej” części cząsteczki silnie zmieniają, którą izoformę preferuje wiązać.
Od algorytmów do praktycznych narzędzi projektowania leków
Aby uczynić podejście dostępnym, autorzy zapakowali trzy modele SVM, warstwę niepewności i silnik kontrfaktyczny do narzędzia graficznego o nazwie CAInsight. Użytkownik może podać tekstową reprezentację cząsteczki i jednym kliknięciem uzyskać przewidywaną aktywność wobec hCA II, IX i XII, oszacowanie wiarygodności każdego przewidywania oraz proponowane modyfikacje strukturalne, które mogą zwiększyć lub zmniejszyć aktywność. Choć modele skupiają się na klasyfikowaniu cząsteczek jako aktywne lub nieaktywne, a nie na przewidywaniu dokładnej mocy działania czy selektywności w jednym kroku, już odtwarzają znane zachowania rzeczywistych kandydatów na leki i wykrywają subtelne zmiany strukturalne. Autorzy zauważają, że większe i bardziej jednorodne zbiory danych oraz głębsza analiza wyboru progów aktywności mogłyby jeszcze poprawić wydajność.
Co to oznacza dla przyszłych leków przeciwnowotworowych
Mówiąc prosto, praca ta pokazuje, że starannie zbudowane i dobrze wyjaśnione modele uczenia maszynowego mogą pomóc chemikom projektować leki przeciwnowotworowe, które lepiej rozróżniają podobne cele enzymatyczne. Łącząc solidne statystyki, oszacowania niepewności i intuicyjne przykłady „co jeśli”, ramy te nie tylko przewidują, które cząsteczki najprawdopodobniej zadziałają, lecz także sugerują dlaczego. Tego rodzaju przejrzysta sztuczna inteligencja może przyspieszyć przesiewy wirtualne, wspierać generatywne projektowanie nowych związków i zmniejszyć ciężar prób i błędów w laboratorium, ostatecznie wspomagając odkrywanie bardziej selektywnych i bezpieczniejszych terapii dla pacjentów.
Cytowanie: Ghamsary, M.S., Rayka, M. & Naghavi, S.S. Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction. Sci Rep 16, 8419 (2026). https://doi.org/10.1038/s41598-026-39771-2
Słowa kluczowe: inhibitory anhydrazy węglanowej, interpretowalne uczenie maszynowe, selektywność leków, predykcja conformalna, wyjaśnienia kontrfaktyczne