Clear Sky Science · pl
Heterozygotyczne mutacje CHAT w złożeniu, mutacja missense i wariant na miejscu splicingu, u dwojga rodzeństwa z wrodzonym zespołem miastenicznym
Kiedy oddychanie zawodzi bez ostrzeżenia
Niektóre dzieci wyglądają na zdrowe przy urodzeniu, a mimo to nagle przestają oddychać podczas drobnych gorączek i wymagają natychmiastowej wentylacji. Dla rodzin te epizody są przerażające i zagadkowe. Badanie opisuje dwoje takich rodzeństwa z Japonii i łączy ich zagrażające życiu napady osłabienia i bezdechu z drobnymi zmianami w jednym genie, który pomaga nerwom komunikować się z mięśniami. Składając do kupy kliniczne wskazówki, sekwencjonowanie genów i komputerowe modelowanie białka, badacze pokazują, w jaki sposób te mutacje prawdopodobnie zaburzają kluczowy enzym i dają lekarzom wyraźniejszy cel diagnostyczny i terapeutyczny.
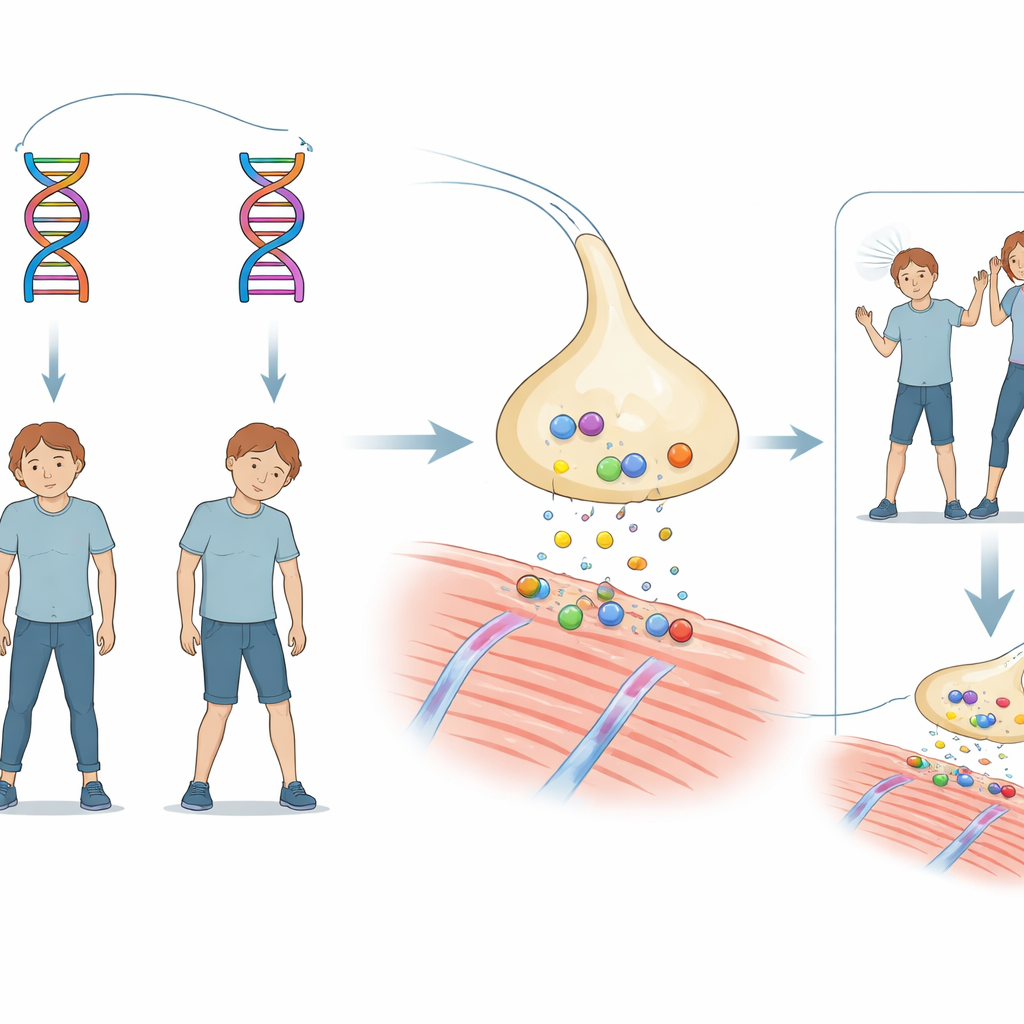
Rodzinna zagadka nagłego osłabienia
Historia koncentruje się na bracie i siostrze, którzy oboje mieli nieco opóźniony rozwój motoryczny w okresie niemowlęcym. Około 18. miesiąca życia każde z nich doświadczyło epizodów bezdechu i utraty przytomności podczas gorączek, na tyle poważnych, że wymagały wentylatora. W miarę dorastania oboje nadal mieli napady opadających powiek i uogólnionego osłabienia mięśni wywoływane przez infekcje, gorączkę lub wysiłek. Badania mózgu były prawidłowe, a powszechne postaci miastenii związane z przeciwciałami zostały wykluczone. Jednak lek nasilający chemiczny sygnał między nerwami a mięśniami wyraźnie poprawiał ich objawy, co wskazywało na rzadką dziedziczną chorobę zwaną wrodzonym zespołem miastenicznym.
Odnalezienie błędnych instrukcji
Aby poszukać przyczyny dziedzicznej, zespół zsekwencjonował wszystkie geny kodujące białka u rodzeństwa i ich rodziców. Stwierdzili, że każde dziecko nosiło dwie różne zmiany w tym samym genie, CHAT, który koduje cholinę acetylotransferazę — enzym wytwarzający acetylocholinę, główny przekaźnik chemiczny wykorzystywany przez nerwy do pobudzania mięśni. Jedna zmiana zmieniała pojedynczy blok budulcowy enzymu (mutacja missense znana jako G411R). Druga znajdowała się na krytycznym granicznym miejscu, gdzie komórka normalnie tnie i łączy segmenty genu podczas tworzenia RNA (mutacja miejsca splicingu oznaczona jako c.752+2T>C). Każdy z rodziców nosił tylko jedną z tych zmian i był zdrowy; tylko dzieci, które odziedziczyły obie zmiany, wykazywały chorobę, co sugeruje, że para mutacji razem osłabia funkcję enzymu.
Badanie, jak ukryte wycięcie zmienia enzym
Ponieważ badacze nie mogli uzyskać wystarczającej ilości naturalnego RNA CHAT z komórek krwi, zastosowali eksperyment „minigenu”. Sklonowali odpowiedni fragment genu do wektora DNA, wprowadzili do hodowanych komórek wersję normalną lub zmutowaną, a następnie zbadali, jak przetwarzany jest RNA. W normalnym konstrukcie RNA zawierał wszystkie oczekiwane segmenty. W mutancie cały odcinek znany jako ekson 5 został pominięty, lecz ogólna ramka odczytu genu pozostała nienaruszona. Oznaczało to, że enzym zostanie zsyntetyzowany, ale będzie brakować krótkiego wewnętrznego odcinka aminokwasów. Porównania ewolucyjne wykazały, że ten brakujący region jest silnie zachowany między gatunkami, co sugeruje, że pełni ważną rolę strukturalną.
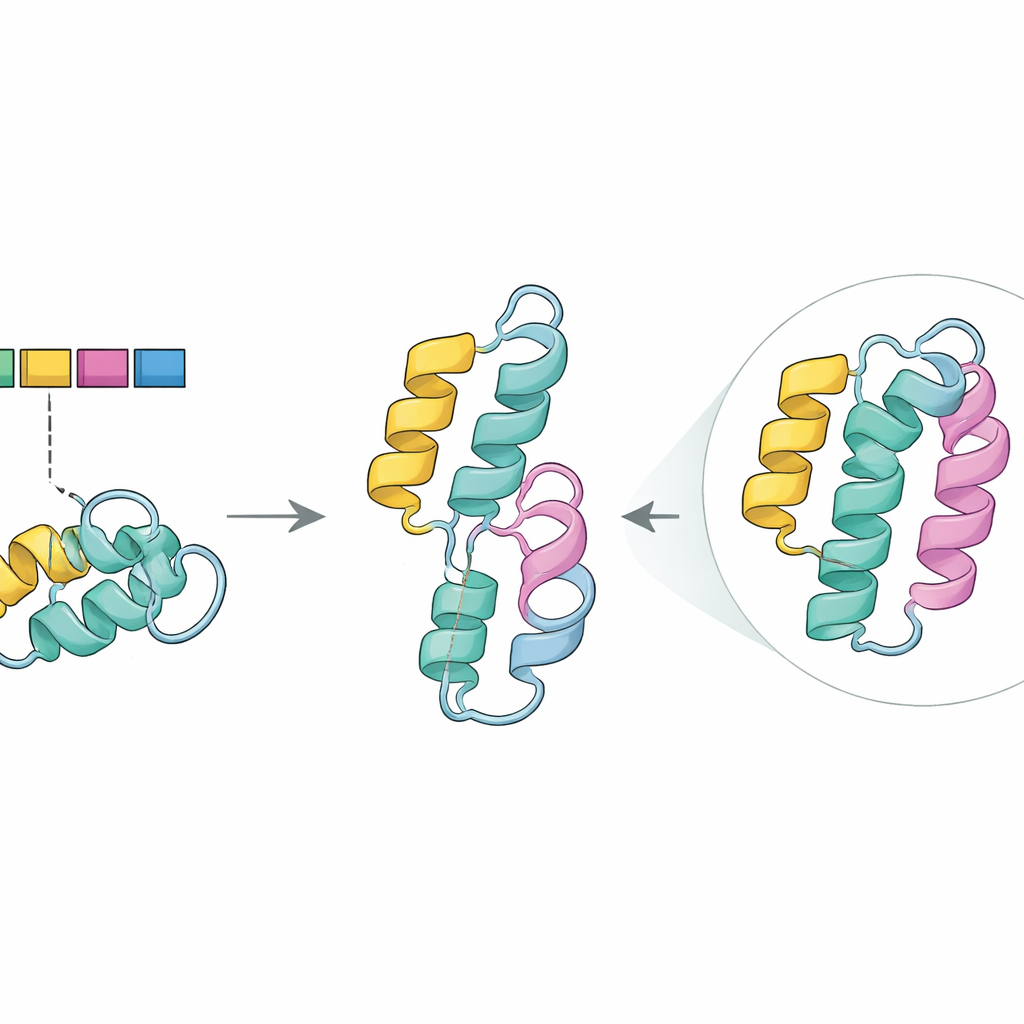
Widzieć uszkodzenie strukturalne in silico
Aby zbadać tę rolę, zespół sięgnął po AlphaFold2, zaawansowany program przewidujący trójwymiarowe kształty białek na podstawie sekwencji. W normalnym enzymie część kodowana przez ekson 5 tworzy jedną ze ściśle upakowanych spiral (alfα‑helisę), które pomagają stabilizować rdzeń białka. W przewidywanej strukturze mutanta ta helisa zniknęła, pozostawiając lukę w regionie znanym z wcześniejszych badań jako kluczowy dla utrzymania stabilności i wspierania efektywnej aktywności chemicznej. W połączeniu z narzędziami komputerowymi wykrywającymi szkodliwe mutacje, wyniki te wspierają hipotezę, że pominięcie eksonu 5, zwłaszcza w zestawieniu z mutacją G411R na drugiej kopii genu, osłabia wydajność enzymu bez jego całkowitego eliminowania — zgodnie z umiarkowanie poważnymi objawami rodzeństwa.
Co to oznacza dla pacjentów i rodzin
Badanie wnioskuje, że kombinacja mutacji missense G411R i nowo zidentyfikowanej mutacji miejsca splicingu w genie CHAT jest bardzo prawdopodobnie przyczyną wrodzonego zespołu miastenicznego u rodzeństwa. Poprzez wykazanie, za pomocą testu minigenu i modelowania strukturalnego, jak zmiana miejsca splicingu usuwa stabilizującą helisę z enzymu, autorzy dostarczają mechanistycznego wyjaśnienia, na którym klinicyści i badacze mogą dalej budować. Dla dotkniętych rodzin taka praca daje więcej niż nazwę: wspiera ukierunkowane leczenie lekami nasilającymi przekazywanie nerwowo‑mięśniowe, kieruje poradnictwem genetycznym przy planowaniu przyszłych ciąż i dodaje istotny nowy przykład do katalogu, jak subtelne zmiany w naszym kodzie genetycznym mogą głęboko wpływać na siłę mięśni i podstawowy akt oddychania.
Cytowanie: Kikuchi, S., Wada, N., Mariya, T. et al. Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome. Sci Rep 16, 9346 (2026). https://doi.org/10.1038/s41598-026-39759-y
Słowa kluczowe: wrodzony zespół miasteniczny, gen CHAT, cholina acetylotransferaza, mutacja miejsca splicingu, złącze nerwowo‑mięśniowe