Clear Sky Science · pl
Modelowanie predykcyjne właściwości fizykochemicznych antybiotyków β-laktamowych za pomocą topologicznych indeksów opartych na wartościach własnych oraz nieliniowych technik regresji
Dlaczego to badanie ma znaczenie
Antybiotyki są filarami współczesnej medycyny, a mimo to bakterie rozwijają oporność szybciej niż odkrywane są nowe leki. Projektowanie lepszych antybiotyków coraz częściej opiera się na modelach komputerowych potrafiących przewidzieć zachowanie kandydata — jak łatwo paruje, jak dużą ma objętość czy jak oddziałuje z wodą i błonami komórkowymi. Artykuł ten bada eleganckie matematycznie podejście do takich przewidywań dla ważnej rodziny leków zwanej antybiotykami β-laktamowymi, wykorzystując narzędzia teorii grafów i statystyki zamiast polegać wyłącznie na kosztownych testach laboratoryjnych.
Przekształcanie cząsteczek w sieci
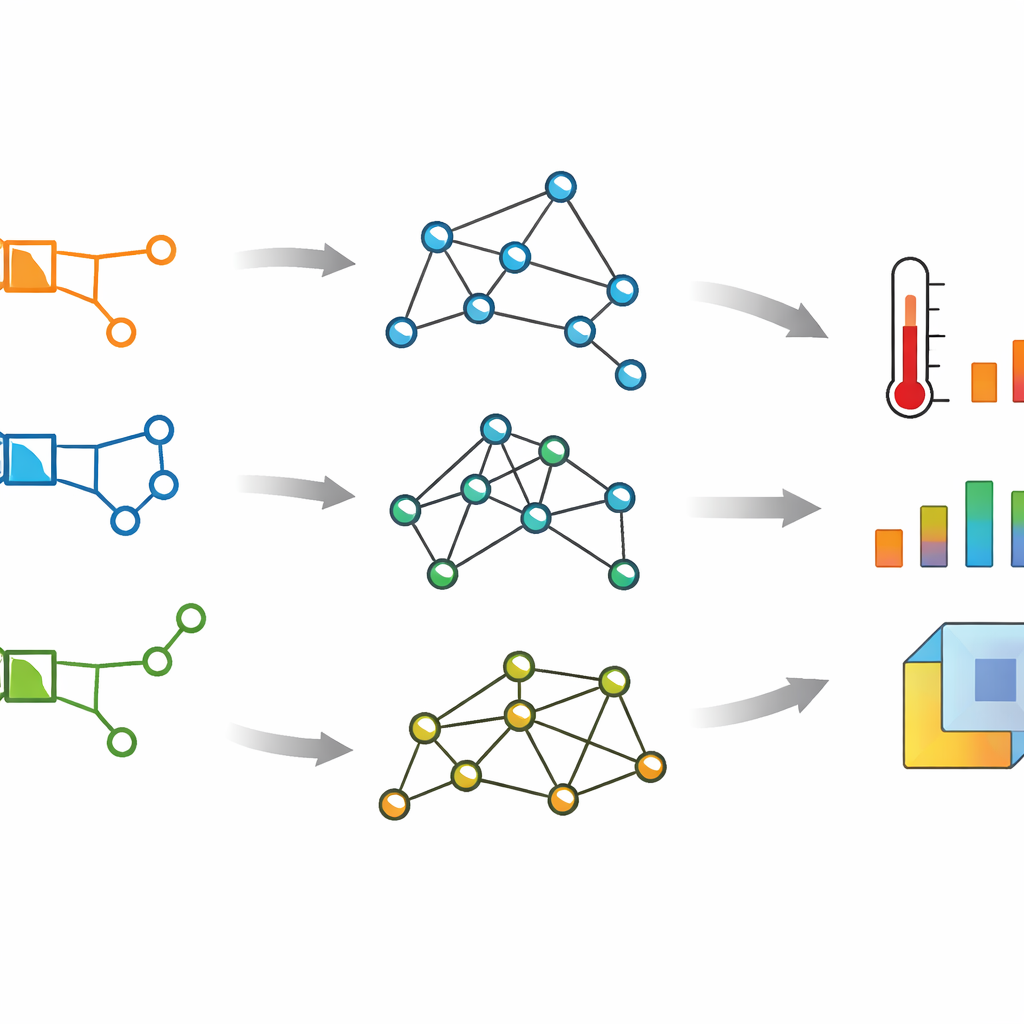
Zamiast postrzegać lek jedynie jako model kulkowo-szprychowy, autorzy traktują każdy antybiotyk β-laktamowy jako sieć: atomy stają się punktami (zwanymi wierzchołkami), a wiązania chemiczne — liniami (krawędziami) łączącymi te punkty. Z tej sieci budują kilka macierzy matematycznych, które uchwytują, jak atomy są połączone, ile wiązań ma każdy atom i jak daleko są od siebie atomy wzdłuż ścieżek wiążących. Te tablice — znane jako macierze sąsiedztwa, Laplace’a, bezznakowa macierz Laplace’a oraz macierz odległości — oferują różne spojrzenia na „kształt” i spójność cząsteczki.
Pomiary ukrytych wzorców w sieci
Gdy te macierze są gotowe, badacze obliczają ich wartości własne, liczby podsumowujące głębokie wzorce strukturalne w sieci. Na podstawie tych wartości własnych konstruują zbiór liczbowych ocen zwanych deskryptorami spektralnymi, o nazwach takich jak energia macierzy sąsiedztwa, algebraiczna spójność czy energia macierzy odległości. Każdy deskryptor łączy informacje z całego grafu cząsteczki, uwzględniając zarówno lokalne szczegóły wokół atomów, jak i globalną architekturę cząsteczki. Ponieważ antybiotyki β-laktamowe mogą różnić się subtelnie w systemach pierścieniowych i łańcuchach bocznych, takie wrażliwe, obejmujące całą cząsteczkę miary są przydatne do wiązania struktury z właściwościami.
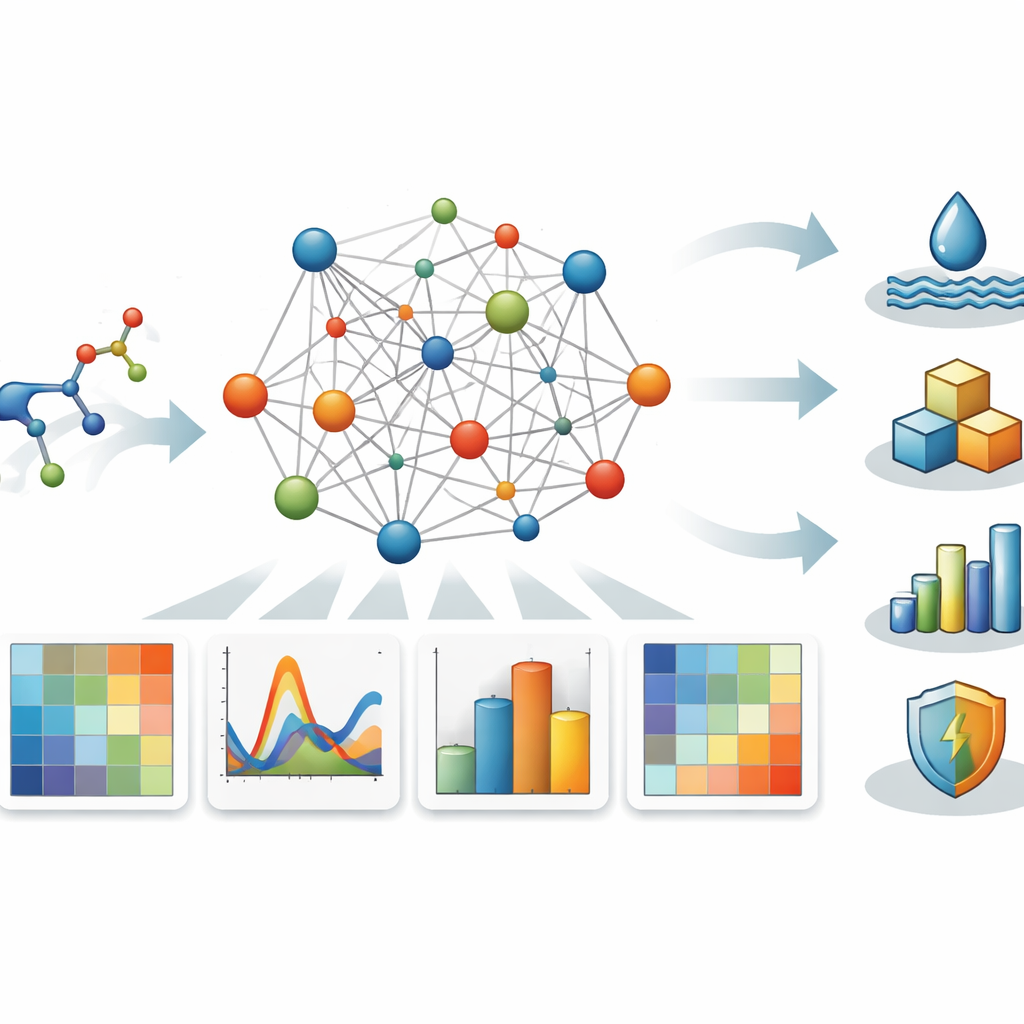
Łączenie ocen struktury z właściwościami codziennego znaczenia
Badanie koncentruje się na siedmiu klinicznie istotnych związkach β-laktamowych, w tym znanych lekach takich jak amoksycylina i imipenem, dobranych tak, by obejmowały różne rozmiary i wzory łańcuchów bocznych. Dla każdego leku zespół zebrał dane eksperymentalne dotyczące praktycznych właściwości fizykochemicznych: temperatury wrzenia, objętości molowej, tego, jak silnie związek załamuje światło (współczynnik załamania molowy), udziału powierzchni polarnej, łatwości zniekształcenia elektronów (polaryzowalność) oraz napięcia powierzchniowego. Następnie przetestowali, jak dobrze każdy pojedynczy deskryptor spektralny potrafi przewidzieć daną właściwość, dopasowując trzy typy nieliniowych zależności — kwadratową, logarytmiczną i potęgową — przy użyciu standardowego oprogramowania statystycznego.
Jak dobrze działają przewidywania?
Wyniki pokazują, że kilka deskryptorów koreluje silnie z właściwościami głównie rządzonymi przez rozmiar cząsteczki i gęstość połączeń atomów. Na przykład algebraiczna spójność, energia bezznakowej macierzy Laplace’a i energia macierzy odległości często wyróżniają się jako szczególnie informatywne. Równania kwadratowe, pozwalające na prostą zależność zakrzywioną między deskryptorem a właściwością, zwykle wypadają nieco lepiej niż formuły logarytmiczne czy potęgowe, dając wyższe współczynniki determinacji i niższe błędy prognozy. Sugeruje to, że związek między strukturą sieciową cząsteczki a jej właściwościami makroskopowymi jest często łagodnie zakrzywiony, a nie liniowy.
Gdzie podejście zawodzi
Modelowanie jest mniej udane dla właściwości, które silnie zależą od rozkładu elektronów na powierzchni cząsteczki i od sposobu tworzenia specyficznych interakcji, takich jak wiązania wodorowe. Powierzchnia polarna czy napięcie powierzchniowe wykazują np. większe rozproszenie między wartościami przewidywanymi a zmierzonymi. Ponieważ stosowane tutaj deskryptory grafowe koncentrują się tylko na tym, które atomy są połączone i jak daleko są od siebie, nie kodują one wprost szczegółowych efektów elektronowych ani kierunkowych interakcji z otoczeniem. Ograniczenie to wynika ze zwięzłości reprezentacji, a nie z nieskuteczności samych metod statystycznych.
Co to oznacza dla przyszłego projektowania antybiotyków
Łącznie badanie pokazuje, że deskryptory grafowe oparte na wartościach własnych oferują zwartą i interpretowalną metodę przewidywania kilku kluczowych właściwości antybiotyków β-laktamowych bez konieczności przeprowadzania pełnego zestawu eksperymentów. Uchwycając ogólny układ i spójność połączeń atomów, te matematyczne miary pomagają prognozować, przy jakiej temperaturze związek wrze, ile zajmuje miejsca oraz jak zachowuje się w masie w kontakcie z otoczeniem. Choć nie mogą jeszcze zastąpić bardziej szczegółowych modeli dla właściwości zależnych od subtelnej struktury elektronowej, stanowią solidną podstawę, którą można łączyć z innymi rodzinami deskryptorów i większymi zbiorami danych. Dla osób niebędących specjalistami wniosek jest taki, że sprytna matematyka zastosowana do planów molekularnych może wspomagać przesiew i optymalizację przyszłych antybiotyków, potencjalnie przyspieszając poszukiwanie leków wyprzedzających oporność bakteryjną.
Cytowanie: Yuvaraj, A., Kalaimurugan, G., Thamizhmaran, R. et al. Predictive modeling for physicochemical properties of \(\beta\)-lactam antibiotics through eigenvalue based topological indices and non linear regression techniques. Sci Rep 16, 9389 (2026). https://doi.org/10.1038/s41598-026-39436-0
Słowa kluczowe: antybiotyki β-laktamowe, modelowanie QSPR, deskryptory teorii grafów, właściwości fizykochemiczne, projektowanie leków