Clear Sky Science · pl
Model analitycznego uczenia napędzanego genami dla dokładnej diagnozy raka piersi
Dlaczego to badanie ma znaczenie dla pacjentów i rodzin
Rak piersi jest obecnie najczęściej rozpoznawanym nowotworem u kobiet na świecie, a pacjentki, które na papierze wyglądają na chore w podobny sposób, mogą mieć bardzo różne rokowania. To badanie pokazuje, jak wzorce w tysiącach genów, połączone ze starannie zaprojektowanym systemem sztucznej inteligencji, mogą pomóc lekarzom bardziej wiarygodnie określić, kto ma raka i jak poważna może być choroba — wykorzystując wyłącznie rzeczywiste dane pacjentów i zwartą grupę kluczowych genów.
Od wielu czynników ryzyka do języka genów
Ryzyko raka piersi kształtują liczne czynniki: odziedziczone zmiany genetyczne, hormony, masa ciała, styl życia i inne. Gdy pojawi się nowotwór, jego zachowanie determinują geny, które są włączone lub wyłączone w każdej tkance guza. Nowoczesne sekwencjonowanie może mierzyć aktywność dziesiątek tysięcy genów jednocześnie, ale przekształcenie tego oceanu liczb w jasne odpowiedzi „tak–nie” dla diagnostyki i rokowania jest trudne. Tradycyjne metody komputerowe często analizują geny pojedynczo i mogą przeoczyć sposób, w jaki grupy genów działają razem, albo wypadają dobrze tylko na jednym zbiorze danych i zawodzą przy testach na innych.
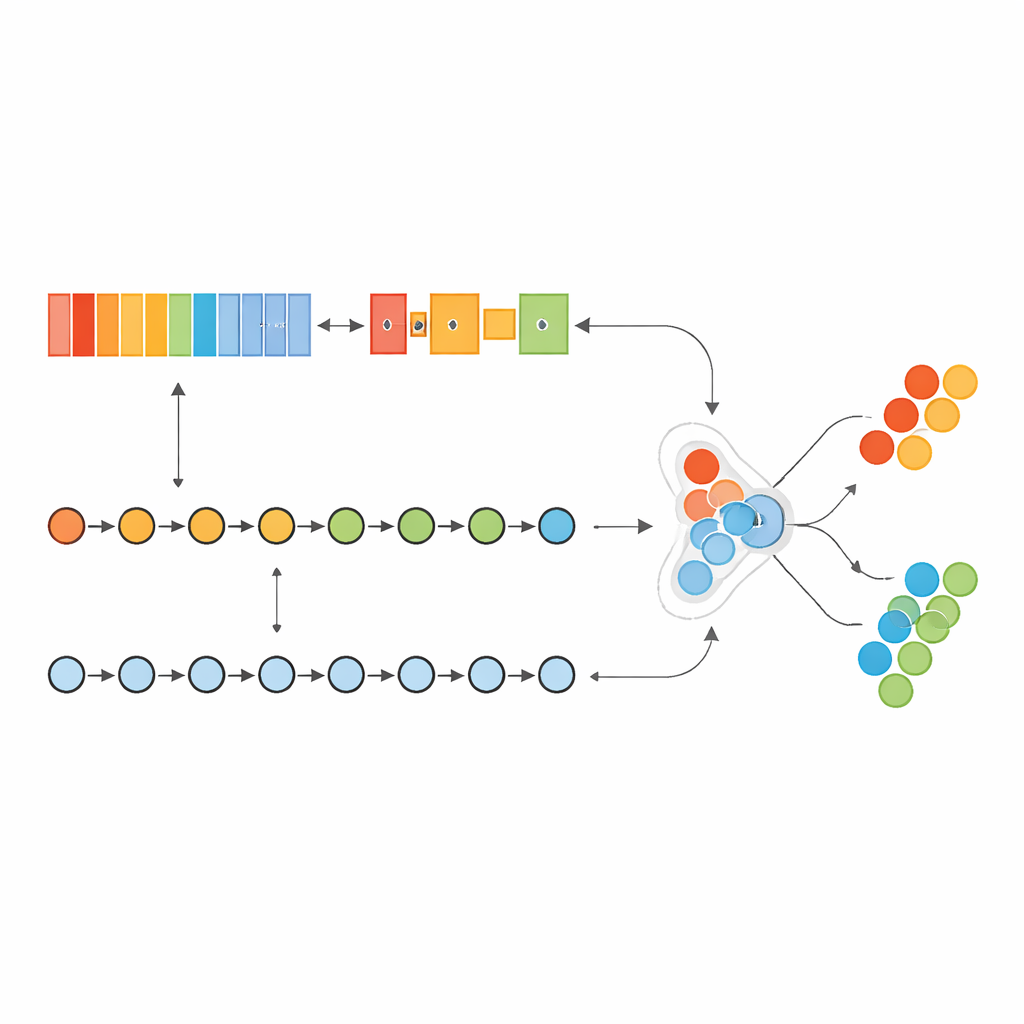
Nauka modelu z podwójnym „mózgiem” czytania wzorców genów
Autorzy zbudowali „hybrydowy” model uczenia głębokiego, działający nieco jak dwa wyspecjalizowane mózgi współpracujące ze sobą. Jedna część, inspirowana analizą obrazów, przeskanowuje uporządkowaną listę genów, aby wykryć lokalne wzorce — klastry genów, których wspólna aktywność sygnalizuje raka. Druga część traktuje te same geny jako sekwencję, ucząc się, jak wczesne „geny napędowe” i późniejsze „geny downstream” wpływają na siebie nawzajem wzdłuż listy. Łącząc te dwa spojrzenia, model potrafi uchwycić zarówno krótkozasięgowe, jak i dalekozasięgowe relacje w genetycznym odcisku guza.
Znalezienie stabilnego rdzenia genów sygnałowych
Zamiast wprowadzać do modelu wszystkie 17 815 zmierzonych genów, zespół zaprojektował rygorystyczny, "wolny od przecieku" pipeline, aby wybrać tylko najbardziej informatywne spośród nich. Korzystając ze standardowej miary korelacji w powtarzanych pętlach walidacyjnych, wielokrotnie porządkowali geny według tego, jak silnie ich aktywność współgrała ze stanem raka. Następnie zachowywali wyłącznie geny, które konsekwentnie pojawiały się na szczycie we wszystkich podziałach treningowych, co dało stabilny sygnaturę 236 genów. Badacze także odwzorowali, jak te geny oddziałują ze sobą, pokazując, że wiele tworzy ściśle połączone sieci związane z wzrostem guza, metabolizmem, odpornością i otoczeniem tkankowym — dowód, że wybrany zestaw odzwierciedla rzeczywistą biologię, a nie losowy szum.
Testowanie modelu
System hybrydowy był trenowany i strojon y na próbkach raka piersi z The Cancer Genome Atlas, a następnie sprawdzany na zupełnie odrębnym zbiorze danych znanym jako METABRIC. Aby poradzić sobie z faktem, że próbki nowotworowe znacznie przewyższają liczbę próbek prawidłowych, autorzy nie tworzyli danych sztucznych; zamiast tego dostosowali, jak bardzo model „zależy” od błędów popełnianych na rzadszej klasie. Po automatycznym poszukiwaniu najlepszych ustawień model osiągnął niemal doskonałe wyniki na głównym zbiorze danych, poprawnie wykrywając prawie wszystkie przypadki raka i praktycznie nie generując fałszywych alarmów. Co ważne, wydajność pozostała bardzo wysoka i stabilna nawet po zastosowaniu modelu do zewnętrznej kohorty METABRIC, co sugeruje, że podejście może uogólniać się poza jedno badanie czy szpital.
Co to oznacza dla przyszłej opieki
Prościej mówiąc, ta praca dostarcza precyzyjnie dostrojony, dwuczęściowy system AI, który odczytuje zwart y kod 236 genów, aby rozróżnić próbki piersi nowotworowe od nienowotworowych z niezwykłą dokładnością i spójnością, nawet w warunkach szumu. Choć obecne badanie dotyczy jedynie aktywności genów i wykorzystuje dane retrospektywne, jego metody tworzą podstawy dla przyszłych narzędzi, które mogłyby łączyć wiele typów danych — takich jak obrazy tkankowe i dodatkowe warstwy molekularne — oraz dostarczać jasnych wyjaśnień, które geny napędzają konkretne przewidywania. Po dalszej walidacji w prospektywnych badaniach klinicznych taki system mógłby stać się uniwersalnym kręgosłupem diagnostyki raka piersi w podejściu precyzyjnej medycyny, pomagając lekarzom dopasować leczenie do genetycznej "sygnatury" guza każdego pacjenta.
Cytowanie: Hesham, F., Abbassy, M.M. & Abdalla, M. Gene driven analytical learning model for accurate breast cancer diagnosis. Sci Rep 16, 8155 (2026). https://doi.org/10.1038/s41598-026-39430-6
Słowa kluczowe: diagnoza raka piersi, ekspresja genów, uczenie głębokie, CNN-BiLSTM, oncologia precyzyjna