Clear Sky Science · pl
CiCLoDS: Wspólne grupowanie komórek i wybór genów dla pojedynczych komórek w transkryptomice przestrzennej
Odnajdywanie sąsiedztw w mieście komórek
Nowoczesne mikroskopy potrafią dziś odczytać, które geny są aktywne w setkach tysięcy komórek, zachowując jednocześnie pozycję każdej komórki w tkance. Ta rewolucja „transkryptomiki przestrzennej” przypomina przekształcenie rozmytej mapy miasta w widok na poziomie ulicy dla każdego domu. Jest jednak pewien problem: te mapy zawierają pomiary dla tysięcy genów na komórkę — znacznie więcej, niż naukowcom łatwo zinterpretować lub zmierzyć w dalszych eksperymentach. W tym badaniu przedstawiono CiCLoDS, nową metodę, która jednocześnie odnajduje biologicznie istotne sąsiedztwa komórkowe i wybiera niewielką, interpretowalną listę genów definiujących te sąsiedztwa.
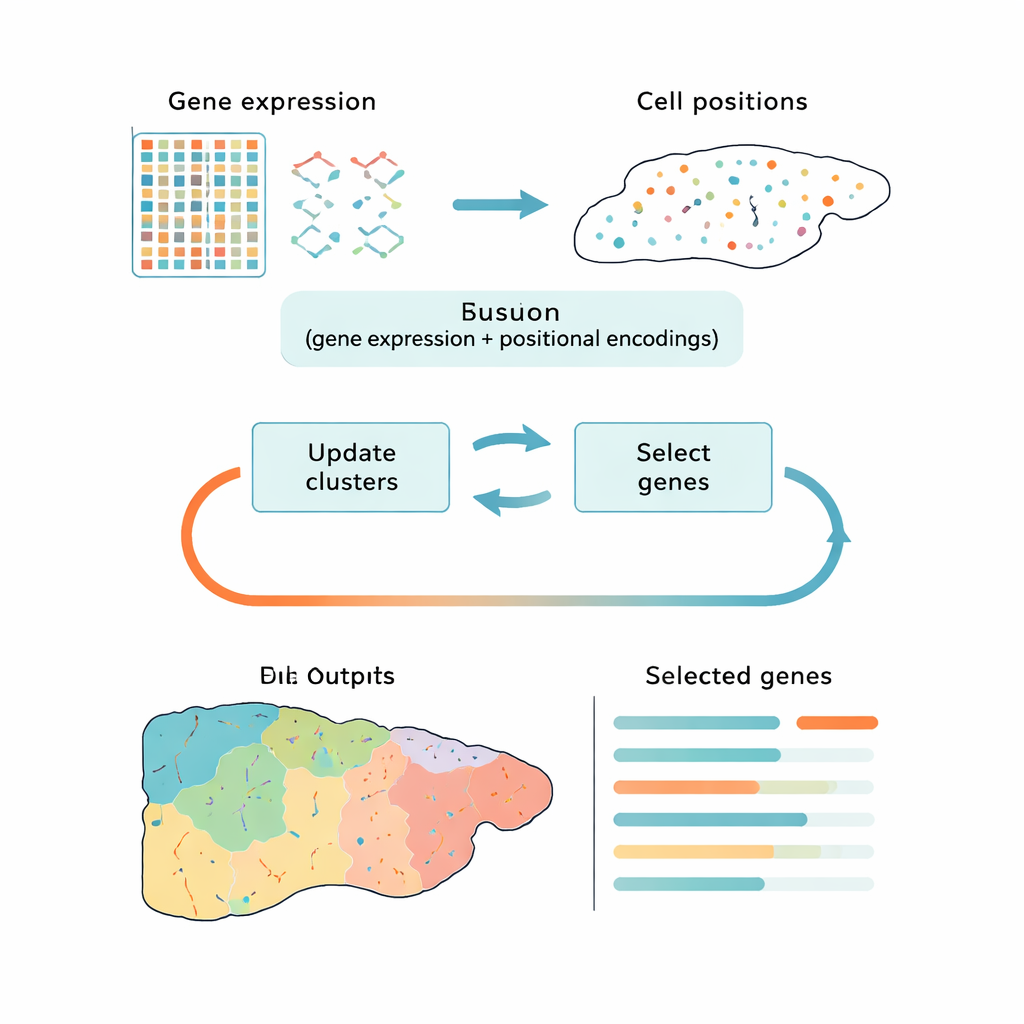
Sprytniejszy sposób na redukcję wielkich danych
Większość obecnych narzędzi radzi sobie z tym wyzwaniem w dwóch odrębnych krokach: najpierw upraszczają dane, potem grupują komórki w klastry. Popularne podejścia, jak analiza głównych składowych (PCA), zachowują ogólną zmienność, ale mogą skupić się na szumie technicznym lub ogólnych sygnałach cyklu komórkowego zamiast na biologicznych różnicach, które mają znaczenie. Inne metody wykorzystują uczenie głębokie do odnajdywania wzorców, lecz działają jak czarne skrzynki i nie wskazują wyraźnie, które geny są najważniejsze. CiCLoDS idzie inną drogą. Traktuje wybór genów i grupowanie jako jeden wspólny problem, pod kontrolą zdefiniowanego przez użytkownika „budżetu” liczby genów, które można zachować. W efekcie pyta: który ograniczony zestaw genów najlepiej wyjaśnia, dlaczego komórki dzielą się na odrębne grupy, biorąc pod uwagę zarówno ich aktywność genową, jak i — gdy są dostępne — położenie w tkance?
Z matematyki do map rzeczywistych tkanek
Autorzy adaptują klasę matematycznie przejrzystych technik zwanych grupowaniem podprzestrzeniowym (subspace clustering) do realiów transkryptomiki przestrzennej, gdzie zbiory danych mogą zawierać ponad milion komórek. CiCLoDS działa na prostej tabeli komórka‑na‑gen, przypisując komórki do klastrów i oceniając każdy gen pod kątem tego, jak bardzo pomaga rozdzielać te klastry. Można też włączyć informacje przestrzenne, dodając pozycyjne „kodowania”, które opisują, gdzie każda komórka znajduje się w tkance, bez zmiany zasadniczej optymalizacji. Na dużych zestawach danych z wątroby myszy i ludzkiego okrężnicy uzyskanych za pomocą platform wysokiej rozdzielczości, CiCLoDS działa w ciągu minut na standardowych komputerach i generuje zwarte panele genów — rzędu kilkudziesięciu do kilkuset genów — które mimo kompresji zachowują bogatą strukturę oryginalnych danych.
Ujawnianie ukrytych stref i naczyń krwionośnych
Zastosowanie CiCLoDS do wątroby myszy pozwoliło sprawdzić, czy metoda potrafi odtworzyć znane wzory „zonacji” — stopniowe zmiany funkcji hepatocytów od jednej strony lobulusa do drugiej. W porównaniu z PCA i wiodącym narzędziem do wyboru genów o nazwie geneBasis, CiCLoDS wygenerował czyściej zdefiniowane strefy przestrzenne z ostrzejszymi granicami i znacznie mniejszą liczbą błędnie przypisanych obszarów, co potwierdziły ilościowe metryki zgodności z mapą odniesienia. Co zaskakujące, przy większym limicie genów CiCLoDS ponownie odkrył grupy hepatocytów przypominające peri‑portalne i peri‑centralne, które bardzo dobrze odpowiadały ekspercko zdefiniowanym klastrom odniesienia, mimo że metoda nie znała kluczowego markera AXIN2 ani nie otrzymała jawnych współrzędnych przestrzennych. Po dodaniu kodowań przestrzennych CiCLoDS również wyselekcjonował panele genów wzbogacone o funkcje związane z powierzchnią komórkową i naczyniami oraz potrafił precyzyjnie odróżnić prawdziwe naczynia krwionośne od artefaktów obrazowania — coś, z czym prostsze metody mierzyły się albo ponosząc porażkę, albo osiągając to tylko dzięki doraźnym modyfikacjom.
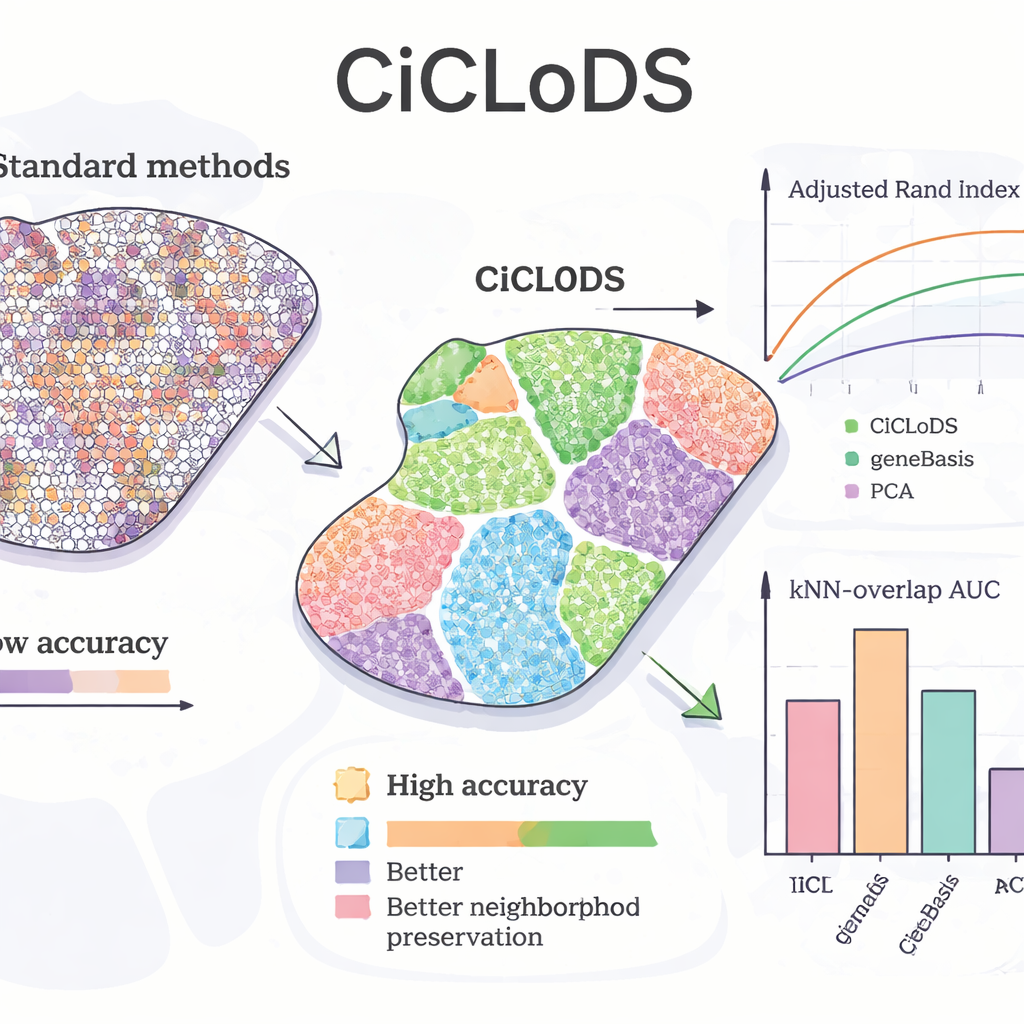
Uogólnianie na różne mózgi i wzmacnianie innych metod
Aby sprawdzić, czy CiCLoDS sprawdza się w bardzo różnych tkankach i u różnych osób, autorzy przeanalizowali próbki kory przedczołowej grzbietowo‑bocznej od trzech dawców. W tych danych CiCLoDS wypadł porównywalnie lub lepiej niż wyspecjalizowane metody przestrzenne, takie jak BayesCafe i BayesSpace, szczególnie na trudnej próbce, gdzie pozostałe narzędzia miały trudności. Badanie podkreśla też zastosowanie „hybrydowe”: uruchomienie CiCLoDS najpierw, by uzyskać stabilne klastry, a następnie przekazanie ich do BayesSpace. Taka strategia warm‑start zwiększyła ogólną dokładność i wygenerowała wzory warstw mózgowych najlepiej zgodne z adnotacjami ekspertów, pokazując, że CiCLoDS może działać samodzielnie i jednocześnie poprawiać niezawodność modeli probabilistycznych wykorzystywanych dalej.
Dlaczego to ma znaczenie dla biologii i medycyny
Dla osób niezajmujących się specjalistycznie najważniejszy wniosek jest taki, że CiCLoDS przekształca przytłaczające mapy komórkowe w zwięzłe, biologicznie sensowne streszczenia. Zamiast pracować z tysiącami zaszumionych pomiarów, badacze otrzymują zarządzalną listę genów i wyraźne, przestrzenne klastry odzwierciedlające prawdziwą organizację tkanki — strefy metaboliczne w wątrobie, naczynia krwionośne i ich nisze oraz warstwowe struktury w mózgu. Ponieważ budżet genowy kontroluje użytkownik, a obliczenia są lekkie, CiCLoDS może pomóc zaprojektować ukierunkowane panele genów do przyszłych eksperymentów, ukierunkować interpretację złożonych zestawów danych przestrzennych i dostarczyć solidnych punktów wyjścia do bardziej złożonego modelowania. W erze, w której wąskim gardłem nie jest już zbieranie danych, lecz ich zrozumienie, narzędzia takie jak CiCLoDS obiecują uczynić wielowymiarowe mapy tkanek zarówno praktycznymi, jak i pouczającymi.
Cytowanie: Wang, N., He, Y., Ray, E. et al. CiCLoDS: Joint cell clustering and gene selection for single-cell spatial transcriptomics. Sci Rep 16, 5356 (2026). https://doi.org/10.1038/s41598-026-39168-1
Słowa kluczowe: transkryptomika przestrzenna, grupowanie komórek, dobór panelu genów, architektura tkanki, analiza pojedynczych komórek