Clear Sky Science · pl
DeepRetro odkrywa retrosyntetyczne ścieżki poprzez iteracyjne rozumowanie dużych modeli językowych
Dlaczego mądrzejsza chemia ma znaczenie
Wiele z dzisiejszych najważniejszych leków i materiałów zaczyna się od złożonych, trudnych do otrzymania cząsteczek. Planowanie, jak zbudować te cząsteczki w laboratorium, przypomina wypracowanie najlepszego sposobu rozebrania, a potem złożenia skomplikowanej maszyny z dostępnych części. Ten etap planowania, nazywany projektowaniem syntezy, często stanowi poważne wąskie gardło w odkrywaniu leków i opracowywaniu zaawansowanych materiałów. W artykule przedstawiono DeepRetro — nowe, otwarte narzędzie, które wykorzystuje duże modele językowe (ten sam typ sztucznej inteligencji, który napędza nowoczesne chatboty) w połączeniu z tradycyjnym oprogramowaniem chemicznym i wiedzą ekspercką, aby projektować realistyczne, krok po kroku procedury syntezy bardzo złożonych cząsteczek.
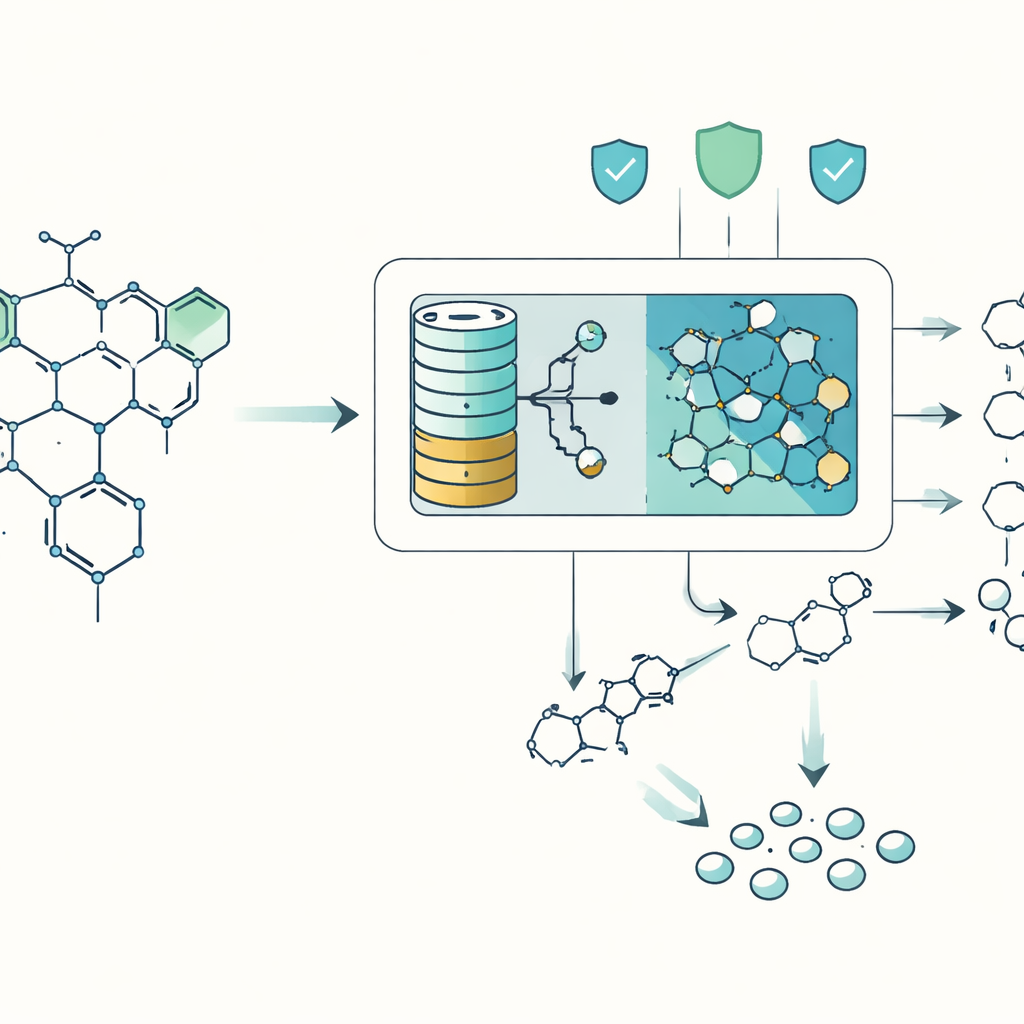
Rozbijanie dużych cząsteczek na przystępne części
Chemicy zwykle planują syntezę, idąc wstecz od docelowej cząsteczki, „odpadając” ją mentalnie na prostsze fragmenty, które można kupić lub przygotować. Komputery pomagają w tym zadaniu od dekad, ale istniejące narzędzia mają trudności, gdy cząsteczki stają się zbyt splątane, egzotyczne lub nieprzypominające niczego z ich baz reakcji. DeepRetro radzi sobie z tym, łącząc dwa światy: szybkie, oparte na regułach silniki stosujące znane wzorce reakcyjne oraz „mózg” oparty na modelu językowym, który może proponować nietypowe, lecz chemicznie sensowne sposoby rozdzielenia cząsteczki. Zamiast prosić AI o wymyślenie całej receptury na raz, DeepRetro prosi je tylko o jeden krok wstecz na raz, a następnie starannie weryfikuje każdą sugestię.
Utrzymywanie AI przy prawdzie
Kluczowym problemem dużych modeli językowych jest to, że mogą "halucynować" — z przekonaniem proponować kroki sprzeczne z podstawową chemią. DeepRetro otacza AI kilkoma warstwami automatycznych kontroli. Każde proponowane pośrednie związek jest testowane pod kątem prostych kryteriów poprawności (na przykład, czy atomy mają odpowiednią liczbę wiązań), prawdopodobnej stabilności oraz wewnętrznej zgodności z resztą reakcji. Sugestie, które nie przejdą tych testów, są odrzucane. Dla propozycji zaakceptowanych system uruchamia następnie bardziej tradycyjny silnik przeszukiwania, aby sprawdzić, czy znana chemia może połączyć te cegiełki z rzeczywistymi, kupnymi materiałami wyjściowymi. Chemicy mogą także interweniować w dowolnym momencie za pomocą interfejsu graficznego: edytować struktury, ponownie uruchamiać tylko fragment ścieżki lub dodać powszechne grupy ochronne, które czynią wieloetapową chemię praktyczną.
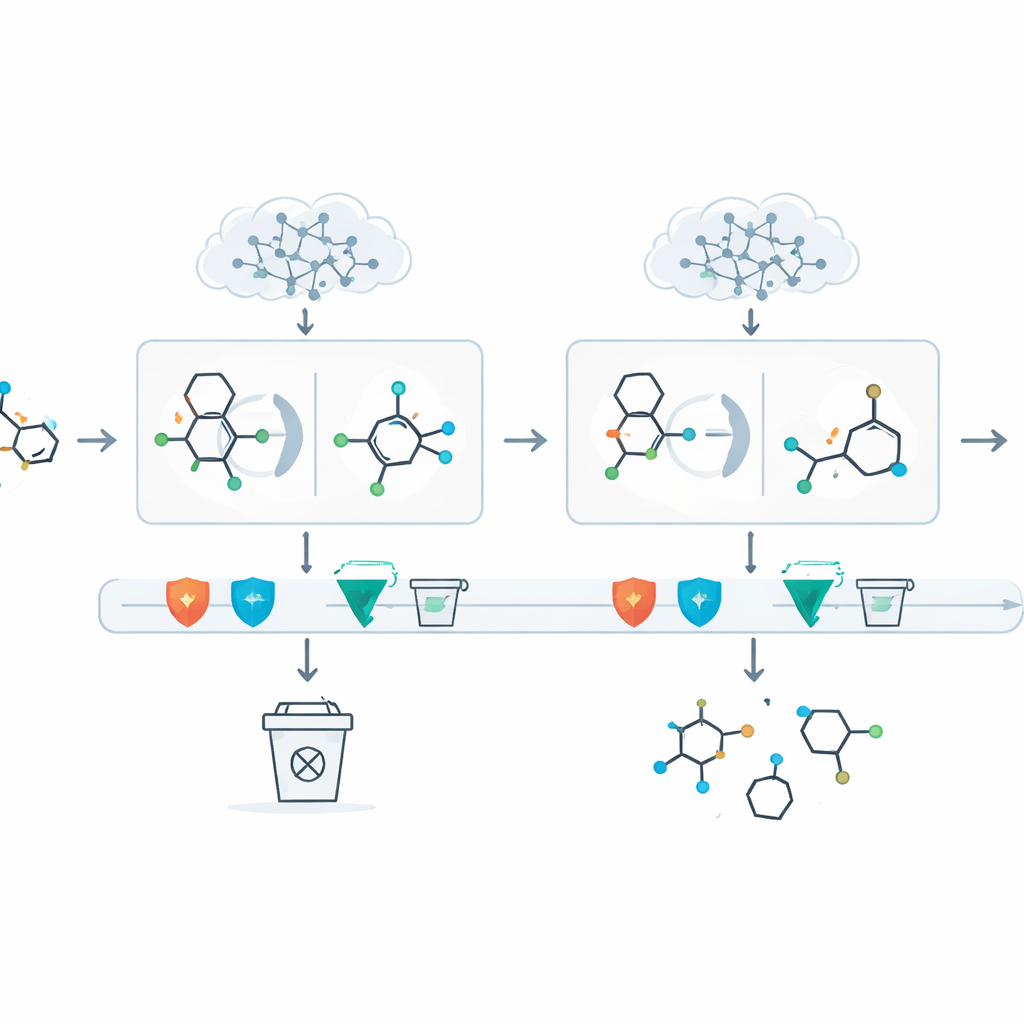
Wystawienie systemu na próbę
Aby sprawdzić, jak dobrze działa DeepRetro, autorzy ocenili go na standardowych zestawach testowych reakcji pochodzących z baz patentowych. Dla przewidywań jednoetapowych — odgadywania, które substraty mogłyby dać dany produkt — system dorównywał lub przewyższał silne istniejące narzędzia w kilku metrykach, szczególnie w poprawnym wskazywaniu głównego prekursoru, nawet gdy składniki uboczne różniły się. W planowaniu wieloetapowym DeepRetro rozwiązał niemal wszystkie cele w dwóch wymagających zestawach testowych, w tym w kolekcji szczególnie trudnych, lekopodobnych cząsteczek, przewyższając wcześniejsze metody będące ówcześnie stanem techniki. Co istotne, testy przeprowadzono w trybie w pełni automatycznym, bez poprawek ludzkich, co pokazuje, że ramy te są odporne nawet zanim zaangażują się doświadczeni chemicy.
Przykłady z realnego świata
Same benchmarki mogą nie oddać tego, na czym chemikom naprawdę zależy: czy proponowana droga wygląda na taką, którą wprawny praktyk rzeczywiście mógłby wypróbować w laboratorium? Autorzy zatem przeanalizowali pięć słynnych, wysoce złożonych produktów naturalnych, w tym antybiotyki erytromycynę B i discodermolide oraz alkaloid reserpinę. W każdym przypadku DeepRetro współpracował z chemikami w iteracyjnym cyklu. AI sugerowało rozłączenia i fragmenty tras; chemicy odrzucali wątpliwe pomysły, korygowali subtelne problemy stereochemiczne i czasami wskazywali systemowi kluczowy pośrednik. W dwóch przypadkach DeepRetro wygenerował pełne plany syntezy, których ogólna strategia nie odpowiadała niczemu, co autorzy znaleźli w literaturze, mimo że pojedyncze reakcje były znane. Wskazuje to, że system potrafi łączyć znaną chemię w autentycznie nowe, globalne rozwiązania.
Obietnice, ograniczenia i dalsze kroki
DeepRetro pokazuje, że duże modele językowe mogą być czymś więcej niż sprytnymi generatorami tekstu; gdy są ściśle nadzorowane i połączone z ugruntowanymi narzędziami, mogą pomagać w nawigacji po ogromnej przestrzeni poszukiwań możliwych syntez chemicznych. Ramy te wciąż mają ograniczenia: ogólnego przeznaczenia modele językowe często proponują niestabilne lub nierealistyczne pośrednie związki, a w pełni automatyczne rozwiązania dla najtrudniejszych cząsteczek pozostają poza zasięgiem bez nadzoru człowieka. Niemniej jednak silne wyniki DeepRetro w standardowych testach, jego sukces w wymagających studiach przypadków oraz otwarte udostępnienie sprawiają, że jest to praktyczny wzorzec dla przyszłych odkryć naukowych wspomaganych przez AI. Dla osób niebędących specjalistami płynie z tego wniosek, że AI przesuwa się od jedynie przewidywania właściwości molekularnych do współprojektowania całych laboratoryjnych procedur, z potencjałem przyspieszenia tworzenia leków i materiałów w nadchodzących latach.
Cytowanie: Sathyanarayana, S.V., Hiremath, S.D., Rahil Kirankumar, S. et al. DeepRetro discovers retrosynthetic pathways through iterative large language model reasoning. Sci Rep 16, 8448 (2026). https://doi.org/10.1038/s41598-026-38821-z
Słowa kluczowe: retrosynteza, duże modele językowe, planowanie syntezy organicznej, odkrywanie leków, chemia obliczeniowa