Clear Sky Science · pl
Obliczeniowa optymalizacja rozpuszczalności domeny kalpainowej DEK1 poprzez zintegrowane modelowanie strukturalne i ukierunkowaną mutagenezę opartą na danych
Dlaczego to, jak zachowują się białka roślinne, ma znaczenie
Wiele białek kontrolujących wzrost roślin to duże, delikatne cząsteczki, które odmawiają rozpuszczenia, gdy naukowcy próbują je badać w laboratorium. Jednym z takich białek jest DEK1, który kształtuje organizmy roślinne zaczynając od pojedynczych komórek. Jednak ponieważ kluczowy fragment DEK1 tworzy agregaty po ekspresji w bakteriach, jego struktura 3D pozostaje nieznana, co spowalnia prace nad zrozumieniem i wykorzystaniem tego białka. W tym badaniu pokazano, jak modelowanie komputerowe i sprytne, oparte na danych projektowanie mogą przeprojektować kłopotliwy region tak, by był bardziej rozpuszczalny, nie naruszając jego ogólnej budowy — oferując ogólną receptę na ujarzmianie trudnych białek.
Skierowanie uwagi na problematyczny fragment kluczowego białka roślinnego
DEK1 to wyjątkowo duże białko osadzone w błonach komórkowych, zakończone regionem enzymatycznym nazywanym domeną kalpainową. Prace genetyczne wykazały, że ta domena jest niezbędna dla prawidłowego rozwoju roślin takich jak mchy i uprawy, lecz jej struktura nigdy nie została rozwiązana eksperymentalnie. Gdy badacze próbują wytworzyć ten rdzeń kalpainowy (nazwany CysPc) w powszechnym gospodarzu Escherichia coli, ma on skłonność do nierozpuszczalności i tworzenia gęstych ciał inkluzyjnych. Utrudnia to niemal całkowicie oczyszczenie go w ilościach i jakości potrzebnej do szczegółowych badań strukturalnych i funkcjonalnych. Autorzy postanowili zatem przeprojektować domenę CysPc tak, by rozpuszczała się łatwiej przy zachowaniu ogólnego kształtu.
Budowanie wiarygodnego modelu 3D od podstaw
Ponieważ nie ma eksperymentalnej struktury tego roślinnego kalpainu, zespół musiał najpierw przewidzieć jego formę 3D. Połączyli kilka najnowocześniejszych narzędzi do przewidywania struktur, w tym AlphaFold2, SWISS-MODEL i I-TASSER, i osadzili te przewidywania względem znanych struktur powiązanych kalpainów ssaczych. Korzystając z podejścia konsensusowego, dopracowali i sprawdzili powstałe modele za pomocą licznych testów jakości oceniających geometrię łańcucha głównego, upakowanie i zgodność ze znanymi wzorcami strukturalnymi. Niezależne kontrole wykazały, że zintegrowany model domeny CysPc był bardziej wiarygodny niż pojedyncze przewidywanie, stanowiąc solidny punkt wyjścia do badania, jak drobne zmiany w sekwencji aminokwasów mogłyby poprawić rozpuszczalność.
Testowanie wirtualnych mutacji w symulowanym rozpuszczalniku
Mając model 3D, autorzy przeprowadzili obszerne symulacje dynamiki molekularnej, w których komputerowo śledzono białko i otaczające je cząsteczki wody w czasie. Skoncentrowali się na resztach na powierzchni białka, które były elastyczne, hydrofobowe lub przewidywano, że sprzyjają agregacji. Kandydackie pozycje mutowano pojedynczo na bardziej przyjazne wodzie aminokwasy, a następnie symulowano każdą wersję przez 200 nanosekund. Dla każdego wariantu mierzono cechy związane z rozpuszczalnością, takie jak obszar powierzchni kontaktujący się z wodą, jak zwarty pozostaje białko oraz jak silnie oscylują atomy. Wiele pojedynczych mutacji umiarkowanie zwiększało kontakt z rozpuszczalnikiem lub wewnętrzne wiązania wodorowe, nie zmieniając jednocześnie ogólnego złożenia, co sugeruje, że podstawowy szkielet CysPc mógł tolerować starannie dobrane podstawienia.
Pozwalając algorytmom przeszukać przestrzeń mutacji
Zmiana pojedynczego resztu rzadko daje dramatyczne korzyści w rozpuszczalności, więc badacze następnie zbadali kombinacje dwóch i trzech mutacji. Wygenerowali bibliotekę wariantów podwójnych i potrójnych zbudowanych z najlepszych pojedynczych mutacji i ponownie symulowali każdy z nich. Aby sprawiedliwie ocenić i uszeregować projekty, zdefiniowali ważony indeks łączący wiele cech z symulacji znanych z korelacji z rozpuszczalnością, nagradzając zwiększoną hydratację i wewnętrzne wiązania, a karząc nadmierną elastyczność. Następnie zastosowali algorytm uczenia ze wzmocnieniem (Proximal Policy Optimization), by poruszać się po ogromnej przestrzeni możliwych potrójnych mutantów i proponować najbardziej obiecujące kombinacje. To poszukiwanie oparte na danych zbiegało się do konkretnego mutanty potrójnego, nazwanego MUT347, jako kandydata pierwszego wyboru.
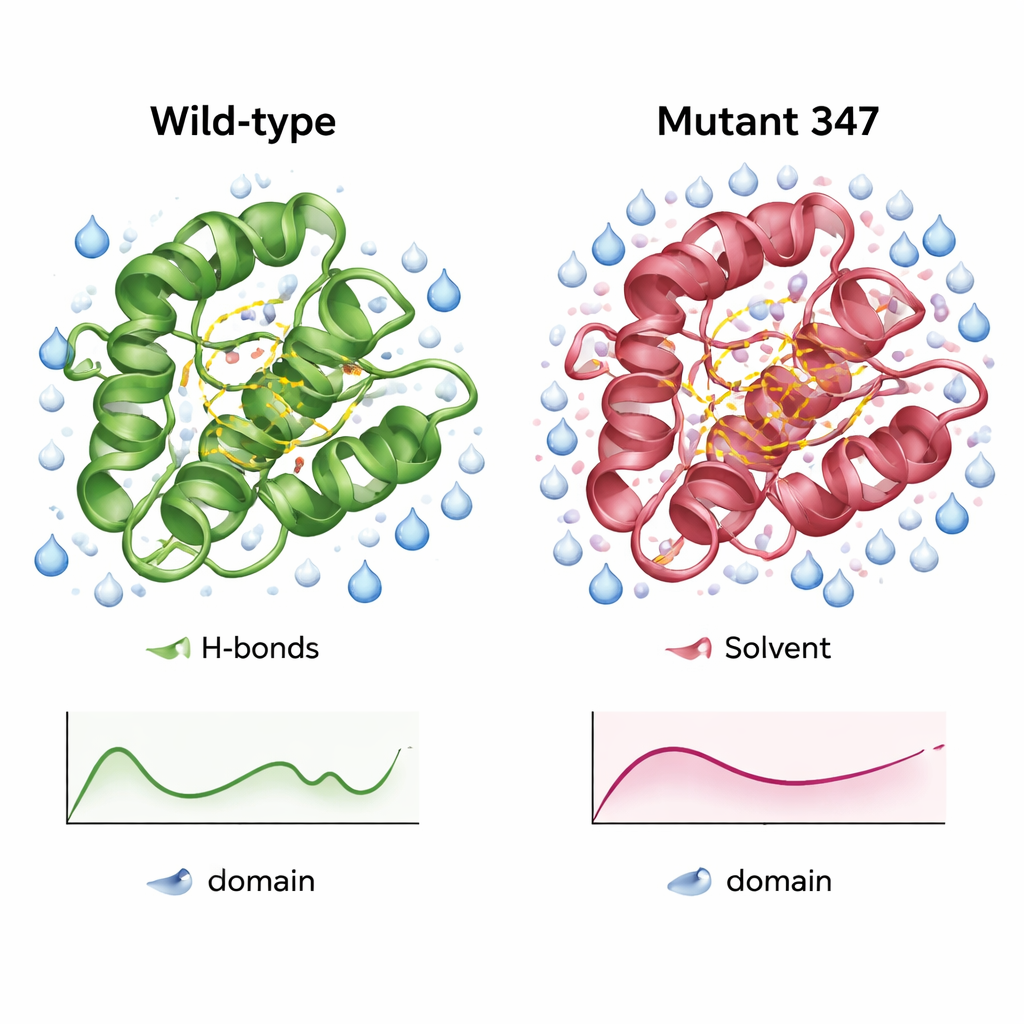
Bardziej zwarta, lepiej nawodniona wersja enzymu
Szczegółowe symulacje dzikiego typu domeny CysPc i MUT347 ujawniły, na czym polegają różnice wariantu inżynieryjnego. MUT347 szybciej osiągał równowagę i wykazywał mniejsze ogólne odchylenia od kształtu początkowego, co wskazuje na większą stabilność strukturalną w roztworze. Jego pętle i końce łańcucha były nieco mniej wiotkie, podczas gdy rdzeń katalityczny zachował pierwotną elastyczność, sugerując zachowanie funkcjonalnie istotnych ruchów. Mutant potrójny miał więcej wewnętrznych wiązań wodorowych i większą powierzchnię dostępną dla wody w kluczowych regionach — oznaki lepiej zorganizowanej i bardziej nawodnionej powierzchni. Przy różnych stężeniach soli i poziomach pH, MUT347 konsekwentnie utrzymywał mniejsze fluktuacje niż białko oryginalne, zachowanie związane ze zmniejszoną skłonnością do tworzenia agregatów.
Co to oznacza dla badań i wykorzystania białek
Dla osób niebędących specjalistami wniosek jest taki, że autorzy stworzyli głównie komputerową procedurę, która przekształca nieporęczny, skłonny do agregacji fragment ważnego białka roślinnego w wersję bardziej rozpuszczalną i łatwiej zachowującą się, bez konieczności uprzedniego poznania jego struktury metodami eksperymentalnymi. Łącząc nowoczesne przewidywanie struktur, długotrwałe symulacje i algorytmy uczące się, które potrafią jednocześnie rozważać wiele decyzji projektowych, zidentyfikowali mutację potrójną przewidywaną jako stabilizującą fałd i korzystniej eksponującą go na wodę. Chociaż prace eksperymentalne są nadal potrzebne, by potwierdzić korzyści w prawdziwych probówkach, ta metoda może być szeroko użyteczna do ratowania innych białek eukariotycznych trudnych w produkcji, ostatecznie pomagając naukowcom udostępniać struktury i funkcje obecnie poza zasięgiem.
Cytowanie: Dabiri, M., Levarski, Z., Struhárňanská, E. et al. Computational optimization of DEK1 calpain domain solubility through integrated structural modelling and data-driven targeted mutagenesis. Sci Rep 16, 7767 (2026). https://doi.org/10.1038/s41598-026-38805-z
Słowa kluczowe: rozpuszczalność białek, obliczeniowa mutageneza, dynamika molekularna, roślinny kalpain DEK1, inżynieria białek