Clear Sky Science · pl
Molekularna charakterystyka recesywnie dziedziczonych ataksji i neuropatii w rodzinach pakistańskich o pokrewieństwie
Dlaczego to ma znaczenie dla rodzin i lekarzy
Problemy z utrzymaniem równowagi, chodzeniem i czuciem w dłoniach i stopach mogą być bardzo upośledzające, zwłaszcza gdy zaczynają się w dzieciństwie i stopniowo nasilają przez całe życie. Dla wielu rodzin, szczególnie w regionach, gdzie dochodzi do małżeństw między kuzynami, objawy te pojawiają się w kilku pokoleniach bez jasnego wyjaśnienia. Badanie to odpowiada na palące pytanie dla takich rodzin w Pakistanie: czy współczesne metody analizy DNA wreszcie ujawnią ukryte przyczyny genetyczne ich ataksji (zaburzeń równowagi i koordynacji) i neuropatii obwodowej (uszkodzenia nerwów kończyn) oraz pomogą lekarzom postawić jaśniejsze rozpoznania i wskazać potencjalne terapie?
Śledzenie choroby dziedzicznej w dużych rodzinach
Naukowcy współpracowali z siedmioma rozbudowanymi pakistańskimi rodzinami, w których kilku członków miało poważne zaburzenia ruchu i funkcji nerwów. U niektórych dominowała ataksja, utrudniająca stabilne chodzenie oraz kontrolę mowy i ruchów oczu. Inni wykazywali klasyczne cechy neuropatii obwodowej, takie jak zanik mięśni rąk i stóp, deformacje stóp i utrata odruchów. W tych rodzinach rodzice byli spokrewnieni, co zwiększało szansę, że dzieci odziedziczą dwie kopie tego samego rzadkiego uszkodzonego genu. Na podstawie próbek krwi od chorych i zdrowych krewnych zespół przeprowadził sekwenowanie eksomu — odczyt niemal wszystkich białkowych fragmentów genomu — aby szukać szkodliwych zmian, które korelowały z występowaniem choroby w drzewach genealogicznych. 
Wykrywanie rzadkich uszkodzonych genów
Po odfiltrowaniu powszechnych i neutralnych różnic w DNA naukowcy znaleźli prawdopodobne warianty chorobotwórcze w pięciu z siedmiu rodzin. Każda z tych rodzin miała własną, specyficzną zmianę genetyczną i wszystkie wykazywały wzorzec recesywny: choroba ujawniała się tylko wtedy, gdy osoba odziedziczyła dwie uszkodzone kopie, po jednej od każdego z rodziców. W jednej rodzinie z początkiem zaburzeń równowagi i trudnościami w mowie w wieku dorosłym przyczyną była rzadka zmiana w genie MFSD8, który pomaga transportować materiały do komórkowych przegrodek recyklingowych zwanych lizosomami. W innej rodzinie szkodliwy wariant w AFG3L2, białku odpowiadającym za utrzymanie mitochondriów — „elektrowni” komórki — wiązał się z młodzieńczą ataksją spastyczną z dystonią (nieprawidłowymi skurczami mięśni). Trzecia rodzina nosiła błąd przesunięcia ramki odczytu (frameshift) w genie SETX, który chroni DNA podczas naprawy i jest już znany jako przyczyna ataksji z apraxią ruchów gałek ocznych, zaburzenia wpływającego również na ruchy oczu.
Bliższe spojrzenie na dziedziczne uszkodzenia nerwów
Dwie kolejne rodziny miały formę choroby Charcot‑Marie‑Tooth (CMT), grupy chorób dziedzicznych uszkadzających długie nerwy do stóp i rąk. W obu przypadkach badacze znaleźli szkodliwe warianty w genie GDAP1, kluczowym dla prawidłowej funkcji mitochondriów w komórkach nerwowych. Jeden wariant w GDAP1 skracał białko i wiązał się z bardzo ciężkim, wczesnym początkiem choroby; inny zamieniał pojedynczy aminokwas w białku i dawał nieco łagodniejszy przebieg. Co godne uwagi, najciężej dotknięty pacjent w jednej z rodzin z CMT był również homozygotyczny dla znanego wariantu chorobotwórczego w drugim genie, MMACHC, który bierze udział w przetwarzaniu witaminy B12 i czasem można go leczyć terapiami opartymi na witaminach. To podwójne obciążenie może tłumaczyć, dlaczego jego objawy były cięższe niż u krewnych, którzy nie mieli wariantu MMACHC.
Gdy poszukiwania w DNA nie wystarczają
Nie każda rodzina przyniosła jasną odpowiedź genetyczną. W dwóch z siedmiu rodzin zespół nie znalazł pojedynczej zmiany w eksomie, która przekonująco odpowiadałaby wzorcowi choroby. W jednym przypadku zidentyfikowano wariant w genie EHHADH, który odpowiadał wzorcowi dziedziczenia, ale przewidywany jest jako łagodny i jest znany z powodowania innej choroby związanej z nerkami, gdy jest zaburzony. W innym przypadku dwaj kuzyni z podobnymi zaburzeniami ruchu okazali się mieć różne podłoża: jeden chłopiec nosił znany szkodliwy wariant w ALS2, który może prowadzić do młodzieńczych form choroby neuronu ruchowego, podczas gdy jego chorzy kuzyni go nie mieli. Te nierozwiązane przypadki sugerują, że ważne mutacje mogą leżeć w częściach genomu, których standardowe sekwenowanie eksomu nie obejmuje, lub że kilka subtelnych czynników genetycznych może współdziałać.
Co to oznacza dla pacjentów i przyszłej opieki
Wyniki pokazują, że nowoczesne narzędzia do analizy DNA mogą ujawnić konkretne geny stojące za złożonymi zaburzeniami nerwów i równowagi, nawet w warunkach o ograniczonych zasobach. Dla pięciu rodzin z jednoznacznymi wynikami praca ta zamienia niejasne etykiety typu „ataksja” czy „neuropatia” w precyzyjne rozpoznania związane z określonymi genami, co może kierować poradnictwem genetycznym, prognozą oraz w niektórych przypadkach wskazywać opcje leczenia, np. terapie związane z witaminą B12 w chorobach związanych z MMACHC. Badanie rozszerza też wiedzę naukowców o tym, jak geny takie jak MFSD8, AFG3L2, SETX, GDAP1, MMACHC i ALS2 kształtują zdrowie komórek nerwowych w mózgu, rdzeniu kręgowym i nerwach obwodowych. W przyszłości potrzebne będą bardziej kompleksowe sekwencje genomu i badania funkcjonalne, aby rozwiązać pozostałe zagadki i przekształcić te genetyczne odkrycia w wcześniejszą diagnozę oraz lepszą opiekę dla dotkniętych chorobą dzieci i dorosłych. 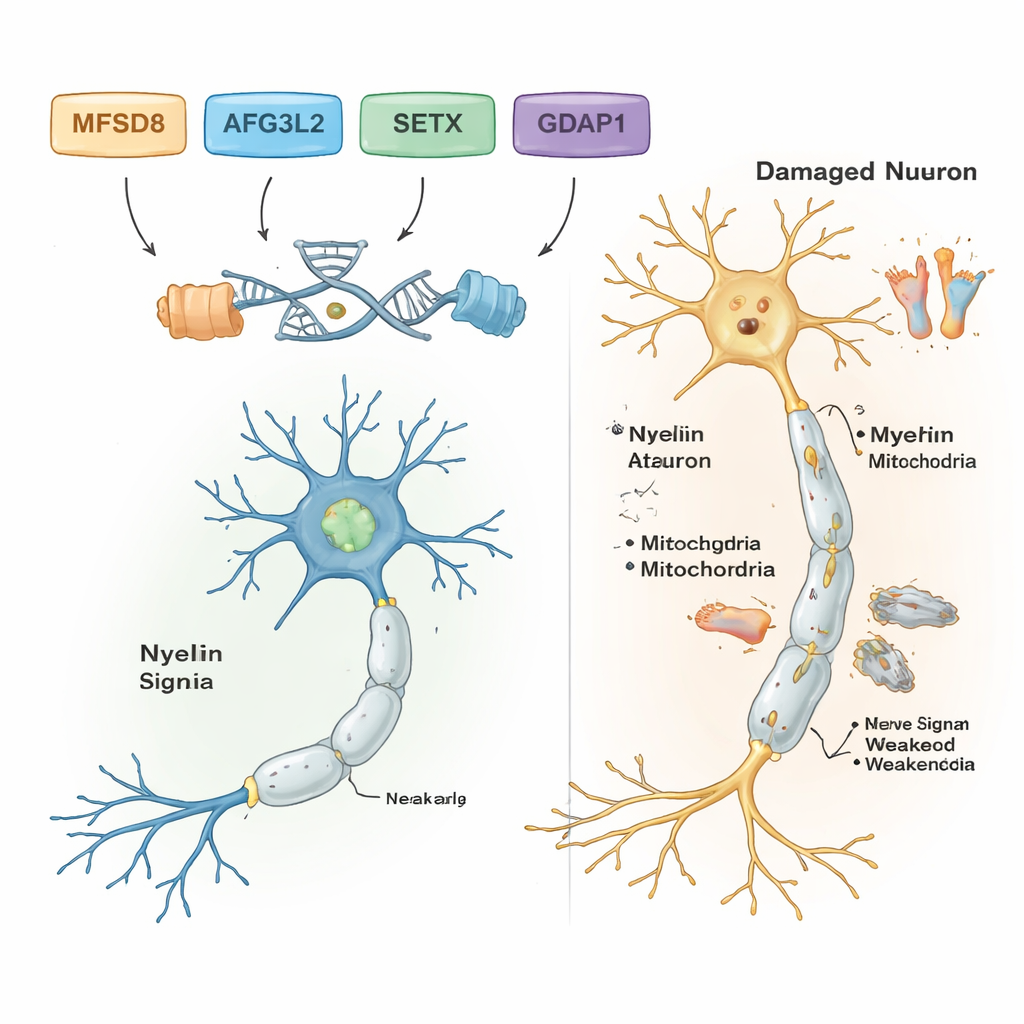
Cytowanie: Aslam, F., Wajid, M., Butt, A.I. et al. Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families. Sci Rep 16, 6529 (2026). https://doi.org/10.1038/s41598-026-37808-0
Słowa kluczowe: ataksja, neuropatia obwodowa, sekwenowanie eksomu, choroba Charcota-Marie-Tootha, diagnostyka genetyczna