Clear Sky Science · pl
Optymalizacja selekcji cech w danych mikroarray nowotworowych za pomocą sterowanego kopcem ramienia ewolucyjnego dla przestrzeni o wysokiej wymiarowości
Dlaczego wybór właściwych genów ma znaczenie
Testy onkologiczne oparte na nowoczesnych technologiach genetycznych potrafią jednocześnie mierzyć dziesiątki tysięcy genów, podczas gdy lekarze często dysponują danymi z zaledwie kilkudziesięciu pacjentów. W tej ogromnej „genowej dżungli” ukryta jest znacznie mniejsza liczba sygnałów, które rzeczywiście odróżniają jeden rodzaj nowotworu od innego lub guz od tkanki zdrowej. W artykule przedstawiono nową metodę inteligentnego przeszukiwania, która automatycznie wyodrębnia te kluczowe geny, mając na celu uczynienie komputerowo wspomaganego diagnozowania nowotworów bardziej dokładnym, szybszym i łatwiejszym do interpretacji.
Za dużo sygnałów, za mało danych
Eksperymenty na mikromacierzach i pokrewnych technologiach pozwalają mierzyć poziomy aktywności tysięcy genów w każdej próbce pacjenta. Jednak liczba próbek zwykle jest bardzo mała, czasem poniżej stu. Wiele odczytów genów jest zaszumionych, redundantnych lub nieistotnych dla rozważanej choroby. Zachowanie ich wszystkich może przytłoczyć algorytmy uczące się, spowolnić obliczenia i prowadzić do mylących modeli, które przyczepiają się do losowych dziwactw zamiast do prawdziwej biologii. Proces redukcji do użytecznego podzbioru nazywa się „selekcją cech” i jest kluczowy, jeśli oczekujemy wiarygodnych predykcji z medycznych danych o wysokiej wymiarowości.
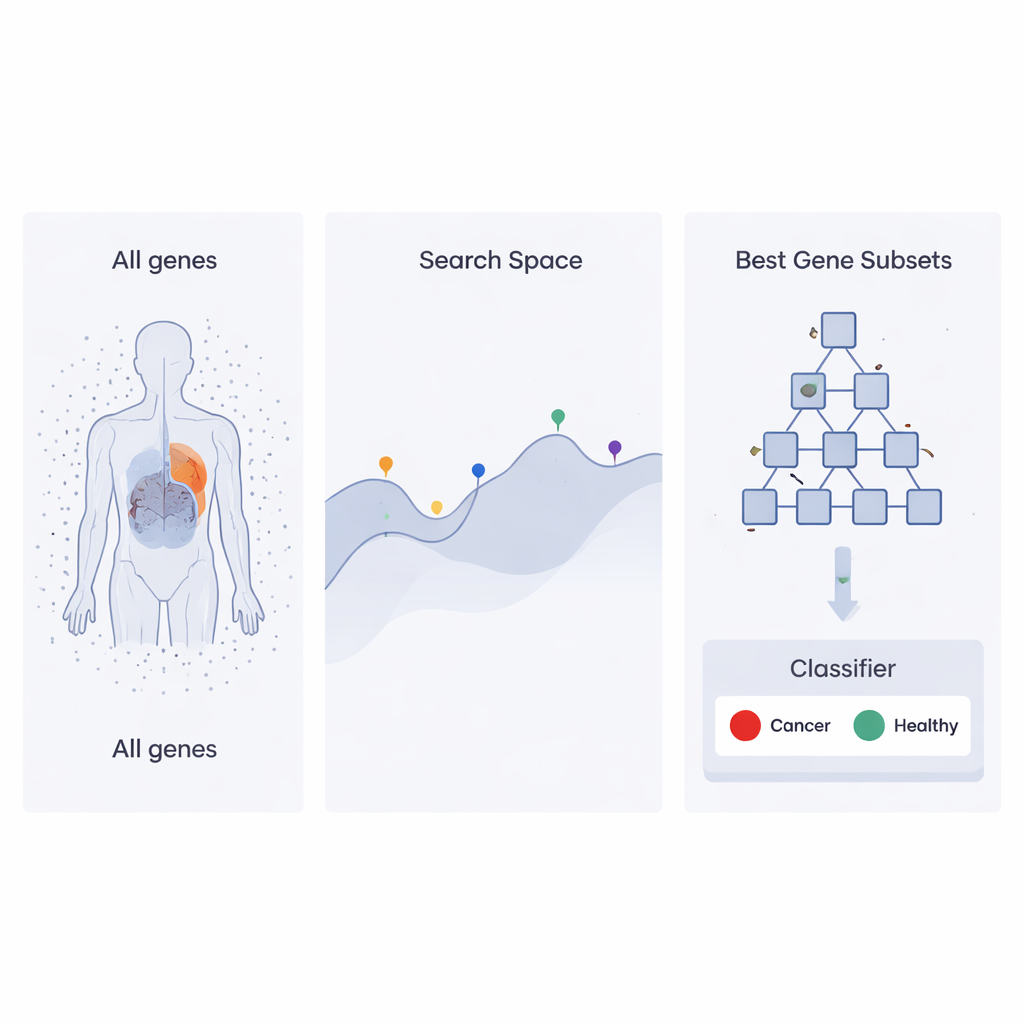
Strategia przeszukiwania inspirowana strukturą korporacyjną
Autorzy rozwijają niedawne podejście optymalizacyjne zwane Heap‑Based Optimizer (HBO), które czerpie pomysły z organizacji pracowników w firmie. Wyobraźmy sobie każdy możliwy zestaw genów jako „pracownika”, którego wydajność oceniana jest na podstawie tego, jak dobrze pomaga klasyfikatorowi rozróżniać próbki nowotworowe od zdrowych. Pracownicy ci są ułożeni w hierarchię, podobną do drabiny korporacyjnej, przy użyciu struktury komputerowej zwanej kopcem. Zestawy genów o wysokiej wydajności zajmują górne pozycje, a słabsze – niższe. W kolejnych rundach niżej sklasyfikowani „pracownicy” dostosowują swoje wybory, kopiując i nieznacznie modyfikując to, co robią ich przełożeni i współpracownicy, stopniowo przesuwając całą organizację w kierunku lepszych rozwiązań.
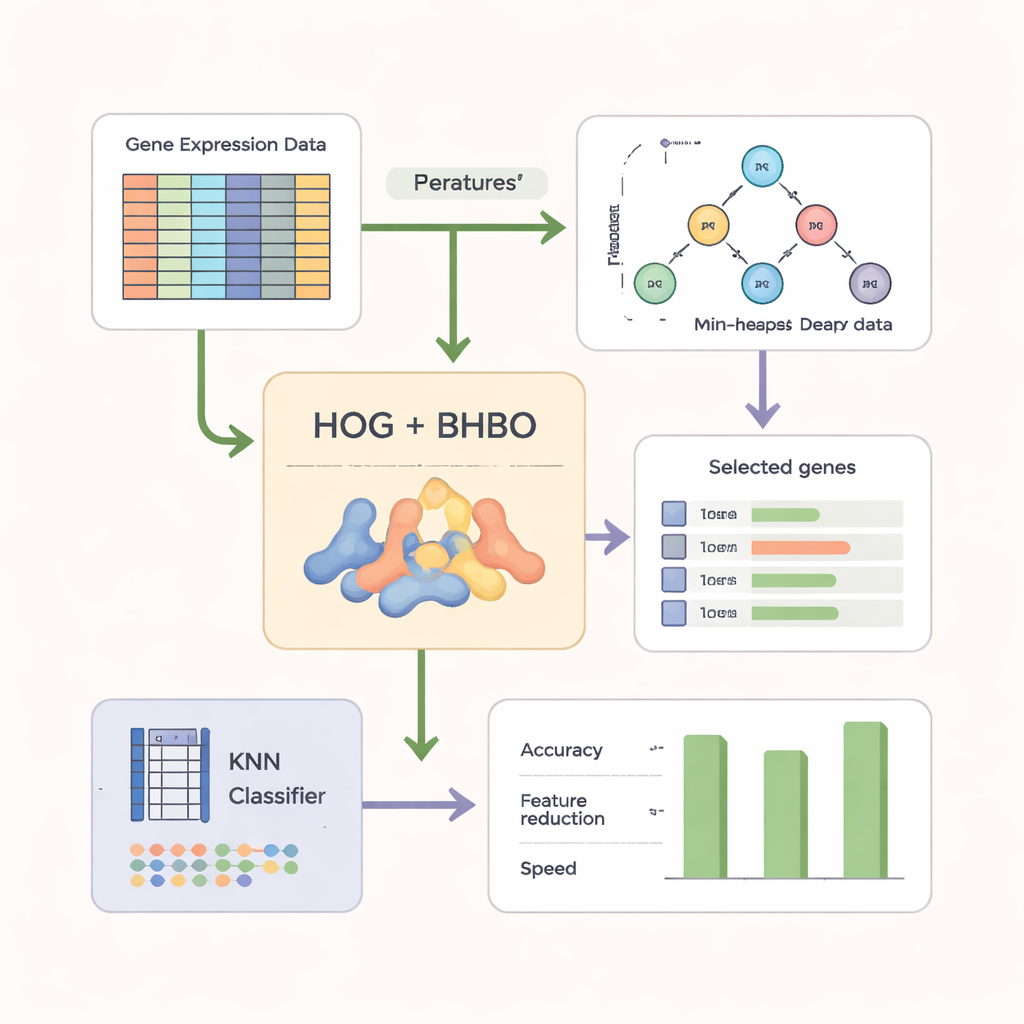
Przekształcanie surowych danych genowych w wyraźniejsze wzorce
Aby uczynić przeszukiwanie skuteczniejszym, autorzy nie polegają wyłącznie na surowych odczytach genów. Najpierw przekształcają dane mikroarray w formę przypominającą obraz i stosują technikę nazwaną Histogram of Oriented Gradients (HOG), szeroko używaną w wizji komputerowej. HOG uchwyca, jak zmieniają się poziomy ekspresji wzdłuż genów, uwydatniając lokalne wzorce zamiast izolowanych pomiarów. Te cechowe wzorce łączy się następnie z oryginalnymi informacjami genowymi. Prosty klasyfikator k‑Nearest Neighbors (KNN) pełni rolę „sędziego”, oceniając każdy kandydacki podzbiór genów na podstawie tego, z jaką dokładnością oznacza nowe próbki, jednocześnie premiując mniejsze, bardziej zwarte zestawy.
Testy na wielu zbiorach danych nowotworowych
Naukowcy ocenili binarną wersję Heap‑Based Optimizer (BHBO) na dziewięciu publicznych zbiorach danych mikroarray dotyczących nowotworów, w tym guzów mózgu, białaczek, raka gruczołu krokowego oraz mieszanych kolekcji guzów z wieloma podtypami. Każdy zbiór zawierał od kilku tysięcy do ponad piętnastu tysięcy mierzonych genów, ale relatywnie niewiele próbek pacjentów. Dla każdego zbioru BHBO uruchamiano wielokrotnie i porównywano z siedmioma dobrze znanymi metodami przeszukiwania, takimi jak algorytmy genetyczne czy optymalizacja rojem cząstek. Zespół mierzył nie tylko dokładność, ale także liczbę zachowanych genów, szybkość zbieżności przeszukiwania oraz stabilność wyników, gdy dane były zaburzane przez symulowany szum, efekty wsadowe i błędy etykiet.
Co osiągnęła nowa metoda
W przekroju dziewięciu zbiorów danych podejście sterowane kopcem osiągnęło średnią dokładność klasyfikacji na poziomie około 95 procent, redukując jednocześnie liczbę genów o ponad 85 procent. Wyraźnie przewyższało konkurencyjne metody dla kilku zbiorów i wykazywało szybszą zbieżność, co oznacza, że szybciej znajdowało dobre zestawy genów w mniejszej liczbie kroków przeszukiwania. Nawet gdy autorzy celowo zniekształcali dane — poprzez dodanie szumu lub odwrócenie etykiet części próbek — wydajność metody spadała tylko nieznacznie i pozostawała lepsza od alternatyw. Testy statystyczne potwierdziły, że uzyskane korzyści mało prawdopodobne były do przypisania przypadkowi.
Co to oznacza dla przyszłych diagnostyk nowotworowych
W praktyce praca ta pokazuje, że starannie zaprojektowana strategia przeszukiwania potrafi przesiać ogromne zbiory genetyczne i odkryć małe, bogate informacyjnie panele genów, które nadal dobrze klasyfikują nowotwory. Dla klinicystów i badaczy takie zwarte zestawy genów są łatwiejsze do biologicznej walidacji, tańsze w pomiarze w testach uzupełniających i bardziej odpowiednie do integracji w narzędziach wspierających decyzje. Chociaż metoda nie odkrywa bezpośrednio nowych leków ani ścieżek biologicznych, zaostrza uwagę na obiecujących markerach genowych, pomagając innym badaniom skupić się na najbardziej informacyjnych sygnałach ukrytych w wysokowymiarowych danych nowotworowych.
Cytowanie: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
Słowa kluczowe: mikromacierz nowotworowa, selekcja cech, optymalizacja metaheurystyczna, biomarkery genowe, eksploracja danych medycznych