Clear Sky Science · pl
Metoda wykrywania wariantów strukturalnych wykorzystująca macierz kontaktów Hi-C i sieci neuronowe
Dlaczego zginanie DNA w 3D ma znaczenie
Nasze DNA zwykle przedstawiane jest jako prosty ciąg liter, ale w każdej komórce zwija się w złożoną trójwymiarową strukturę. Gdy duże fragmenty tego ciągu są usuwane, odwracane lub przemieszczane — zmiany zwane wariacjami strukturalnymi — mogą zaburzać geny i przyczyniać się do powstawania nowotworów. W tym badaniu przedstawiono VarHiCNet, nowy system sztucznej inteligencji, który analizuje mapy fałdowania DNA w 3D i wykrywa te ryzykowne, dużej skali zmiany dokładniej niż istniejące narzędzia, oferując nowe spojrzenie na badanie genomów nowotworowych i innych chorób.
Oglądanie zmian w genomie przez mapy kontaktów 3D
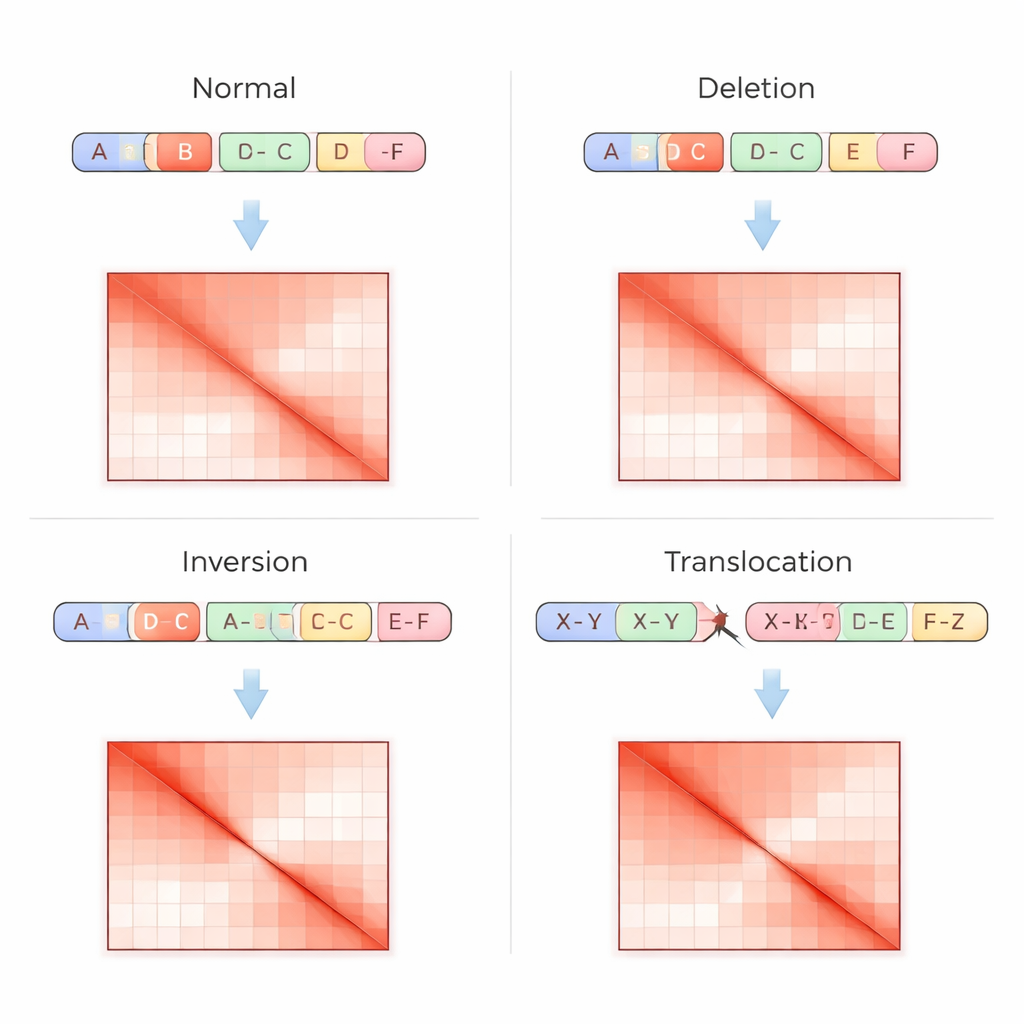
Tradycyjne testy genomiczne odczytują DNA jako liniową sekwencję, co utrudnia wykrywanie skomplikowanych przestawień, zwłaszcza w regionach powtarzalnych albo gdy fragmenty są przestawione bez zmiany liczby kopii. Technika Hi-C podchodzi do problemu inaczej: mierzy, jak często odległe części DNA fizycznie stykają się w jądrze komórkowym, a następnie zapisuje te kontakty jako siatkę, czyli macierz kontaktów, gdzie jaśniejsze pola oznaczają silniejszą interakcję. Wariacje strukturalne pozostawiają w tych macierzach charakterystyczne odciski — na przykład brakujące pasy tam, gdzie region został usunięty, lustrzane wzory przy odwróceniu fragmentu albo ogniska poza przekątną przy złączeniu dwóch chromosomów. VarHiCNet został zaprojektowany, by rozpoznawać te wizualne wzorce automatycznie.
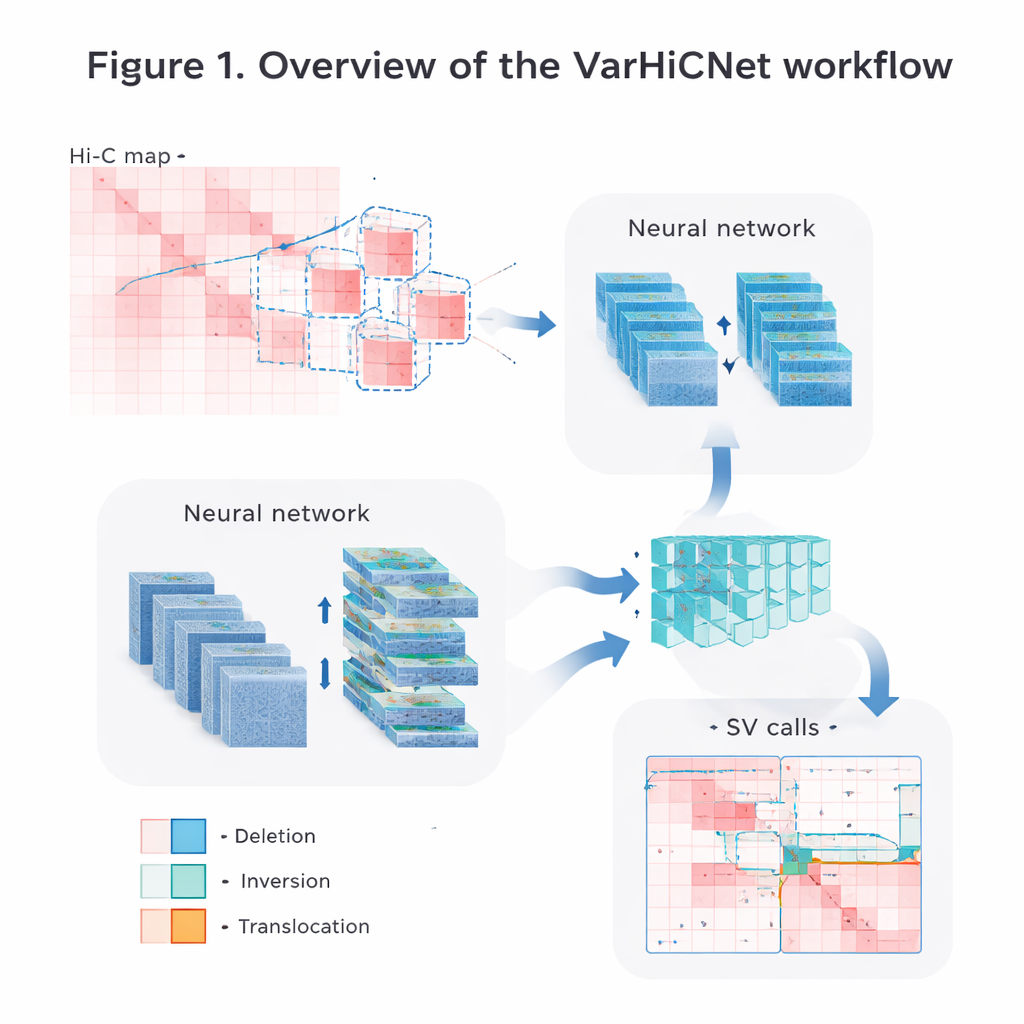
Przekształcanie map genomu w obrazy dla AI
Autorzy zamieniają surowe dane kontaktów Hi-C na obrazy, które systemy widzenia komputerowego mogą łatwo przetwarzać. Najpierw starannie normalizują macierze, aby skorygować naturalny spadek częstości kontaktów wraz z odległością między segmentami DNA, jednocześnie zachowując sygnały zarówno lokalne, jak i dalekozasięgowe. Następnie skanują każdy chromosom nakładając na siebie kwadratowe okna i wycinają wiele mniejszych podmacierzy. Każdą podmacierz skalują do standardowego obrazu kolorowego o rozdzielczości 800 na 800 pikseli, gdzie różne siły kontaktów są mapowane na czerwone odcienie w trzech kanałach kolorów. Taka reprezentacja w formie obrazu pozwala modelowi wykorzystać potężne techniki pierwotnie opracowane do rozpoznawania obiektów na zdjęciach.
Zap借nienie tricków z detekcji obiektów
VarHiCNet traktuje każdy potencjalny wariant strukturalny jak „obiekt” na obrazie. Bazuje na nowoczesnym frameworku do wykrywania obiektów o nazwie RT-DETR, który wykorzystuje kombinację splotowych sieci neuronowych i Transformerów do wyłaniania istotnych regionów. Szkielet ResNet najpierw wydobywa cechy wieloskalowe: płytkie warstwy zachowują drobne szczegóły potrzebne do precyzyjnego określenia miejsc przerwania, podczas gdy głębsze warstwy wychwytują szersze wzorce sygnalizujące duże zdarzenia. Moduł fuzji cech następnie łączy informacje z kilku warstw tak, aby zachować zarówno wskazówki lokalne, jak i globalne. Inny niestandardowy blok, inspirowany przestrzennym agregowaniem piramidalnym, reguluje obszar, jaki model „widzi” naraz, czyniąc go wrażliwym na warianty obejmujące od stosunkowo krótkich po bardzo długie odcinki DNA.
Od kandydatów do precyzyjnych typów wariantów
Gdy VarHiCNet zaproponuje kandydackie regiony na obrazie Hi-C, musi je dopracować do dokładnych miejsc przerwań i konkretnych typów wariantów, takich jak delecje, inwersje, duplikacje czy translokacje. W tym celu system przybliża się do otoczenia każdego przewidywanego przerwania i redukuje jego złożoność za pomocą techniki matematycznej zwanej analizą głównych składowych (PCA), która uwypukla miejsca, w których wzorzec kontaktów zmienia się najostrzej. Te skondensowane reprezentacje są następnie podawane do klasyfikatora opartego na Transformerze, który uczy się subtelnych różnic w lokalnych wzorcach dla poszczególnych kategorii wariantów. Wynikiem jest szczegółowe zgłoszenie każdego zdarzenia: gdzie występuje w genomie i jaki rodzaj zmiany strukturalnej reprezentuje.
Wydajność w różnych liniach komórkowych nowotworów
Naukowcy przetestowali VarHiCNet na danych Hi-C z sześciu różnych ludzkich linii komórkowych nowotworowych, obejmujących nowotwory krwi, piersi, mózgu, nerek, płuc i prostaty. Korzystając z katalogu wariantów strukturalnych o wysokim poziomie ufności jako standardu odniesienia, porównali swoją metodę z kilkoma wiodącymi narzędziami również analizującymi dane Hi-C. Zarówno dla zdarzeń wewnątrzchromosomowych, jak i międzychromosomowych, VarHiCNet zazwyczaj osiągał wyższe lub porównywalne wartości F1, co oznacza lepsze zrównoważenie czułości i dokładności niż inne podejścia. Był szczególnie skuteczny w wykrywaniu zrównoważonych translokacji i inwersji — przestawień, które często pozostawiają niewiele śladów w standardowym sekwencjonowaniu, ale zostawiają wyraźne sygnatury w 3D. Autorzy wykazali także, że ich wybory projektowe, takie jak rozdzielczość obrazu i moduły fuzji cech, konsekwentnie poprawiały wydajność w testach kontrolowanych.
Co to oznacza dla zrozumienia chorób
Mówiąc prościej, VarHiCNet daje naukowcom mądrzejszy sposób „patrzenia” na to, jak genom zwija się w 3D, i wykrywania dużych, związanych z chorobą przestawień, które mogłyby umknąć konwencjonalnemu sekwencjonowaniu. Przekształcając złożone mapy kontaktów w obrazy i stosując nowoczesne sieci wzorowane na metodach widzenia, metoda może wykrywać i klasyfikować wiele rodzajów wariantów strukturalnych z wysoką niezawodnością w różnych typach komórek nowotworowych. Choć nadal ma trudności z bardzo małymi lub silnie splątanymi zmianami i zależy od bogatych danych treningowych, VarHiCNet wskazuje na przyszłość, w której architektura 3D genomu stanie się rutynową częścią odczytu, interpretacji, a ostatecznie ukierunkowanego leczenia zmian genetycznych leżących u podstaw nowotworów i innych chorób.
Cytowanie: Shen, J., Wang, H., Zhai, H. et al. A method for structural variant detection using Hi-C contact matrix and neural networks. Sci Rep 16, 7324 (2026). https://doi.org/10.1038/s41598-026-37678-6
Słowa kluczowe: wariacje strukturalne, Hi-C, uczenie głębokie, genomika onkologiczna, 3D genom