Clear Sky Science · pl
Populacje APOL1 w błonie komórkowej są odporne na szybki rozkład białka
Dlaczego „znikanie” białka nerkowego ma znaczenie
Znaczna część ciężkich chorób nerek u osób o niedawnych afrykańskich przodkach została powiązana z dwoma wariantami jednego genu, APOL1. Naukowcy wciąż jednak mają trudności z wyjaśnieniem, w jaki sposób ten gen uszkadza komórki nerek, nie czyniąc szkody większości nosicieli. Badanie stawia pozornie proste, ale doniosłe pytanie: gdy białko APOL1 zostanie wytworzone w komórce, jak długo się utrzymuje i gdzie jest najbardziej stabilne? Odpowiedzi ujawniają zaskakującą dwubiegunowość — APOL1 jest szybko niszczony wewnątrz komórki, lecz pozostaje uporczywie stabilny, gdy osadzony jest w zewnętrznej powierzchni komórki, co może naprowadzić na kierunki terapii.
Gen ryzyka o podwójnym ostrzu
Gen APOL1 pomaga chronić ludzi przed pewnymi pasożytami — przewaga ewolucyjna, która prawdopodobnie wyjaśnia powszechność wariantów ryzyka G1 i G2 w populacjach afrykańskich. Niestety osoby dziedziczące dwie kopie tych wariantów są obarczone znacznie wyższym ryzykiem chorób nerek zaliczanych do APOL1-zależnych. Wcześniejsze badania wykazały, że gdy poziomy APOL1 rosną — często w odpowiedzi na stan zapalny — białko może stać się toksyczne, szczególnie w wrażliwych komórkach filtrujących nerki, zwanych podocytami. Jednak większość prac skupiała się na tym, co włącza APOL1. Znacznie mniej wiadomo o tym, jak komórki wyłączają je ponownie, na przykład przez rozkład białka.
Śledzenie kruchego białka wewnątrz komórek
Aby zbadać stabilność APOL1, badacze zmodyfikowali linie komórkowe człowieka tak, by produkowały fluorescencyjnie znakowane wersje APOL1 i jego najbliższego krewnego, APOL2. Pozwoliło to obserwować, ile każdego białka gromadziło się lub zanikało w różnych warunkach za pomocą Western blot, mikroskopii i cytometrii przepływowej. Zablokowali główny aparat rozkładu białek w komórce — proteasom — i oddzielnie zatrzymali nową syntezę białek. Przy hamowaniu proteasomów poziomy APOL1 szybko wzrastały, co świadczy, że normalnie jest on szybko rozkładany. Gdy zatrzymano nową syntezę, APOL1 gwałtownie spadał. W wyraźnym kontraście APOL2 prawie nie zmieniał się przy żadnym z tych zabiegów, co ukazało jego większą stabilność. Co istotne, wysoki obrót APOL1 był taki sam dla wersji normalnej (G0) i wariantów ryzyka (G1 i G2) oraz dotyczył kilku naturalnie występujących form APOL1 różniących się unieruchomieniem w błonach.
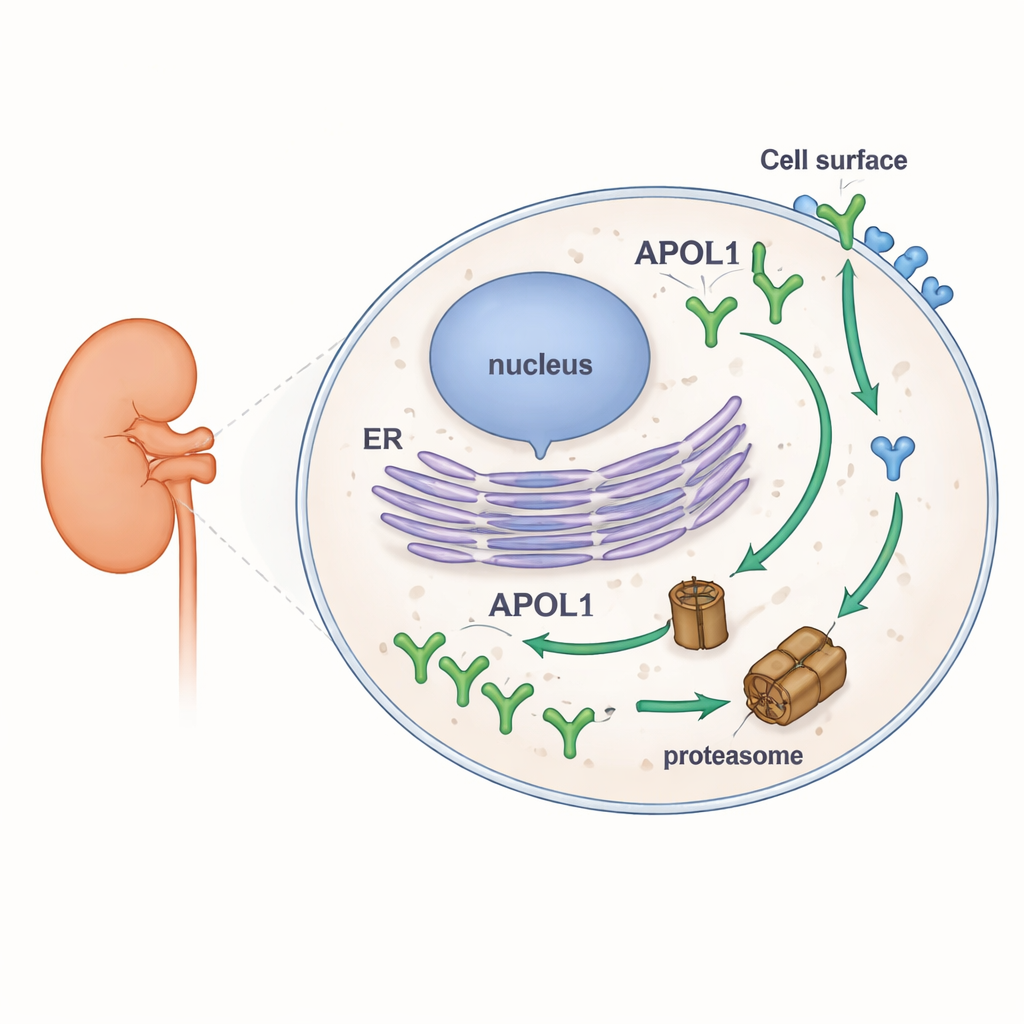
Wskazówki w sekwencji i opowieść o dwóch „sąsiedztwach”
Zanurzając się w strukturę białka, zespół użył narzędzi komputerowych, by przeskanować APOL1 i APOL2 pod kątem luźnych, nieustrukturyzowanych fragmentów zwanych wewnętrznie nieuporządkowanymi regionami. Takie regiony często pełnią rolę sygnałów „zjedz mnie” dla proteasomu. Zidentyfikowali dwa silne kandydatów w APOL1, które były w dużej mierze nieobecne w APOL2. Aby sprawdzić, czy unikatowy koniec N‑terminalny APOL1 przyczynia się do jego kruchości, stworzyli hybrydy: skróconą formę APOL1 pozbawioną pierwszych 59 aminokwasów oraz chimeryczne APOL2 zawierające ten fragment APOL1. Dodanie fragmentu N‑terminalnego APOL1 do APOL2 spowodowało szybszy rozkład APOL2, podczas gdy skrócony APOL1 pozostał niestabilny, co sugeruje, że więcej niż jedna część APOL1 sprzyja szybkiemu rozpadowi. Razem wyniki łączą nietypowe, elastyczne fragmenty APOL1 z jego szybkim obrotem, nie wiążąc tego zachowania wyłącznie z wariantami chorobotwórczymi.
Uparte białko na powierzchni komórki
Najbardziej uderzające odkrycie pojawiło się, gdy autorzy rozróżnili APOL1 znajdujący się wewnątrz komórki od APOL1 na powierzchni komórki. Używając przeciwciał rozpoznających tylko część APOL1 wystawioną na zewnątrz, mierzyli poziomy powierzchniowe oddzielnie od całkowitych. Wewnątrz komórki APOL1 zachowywał się zgodnie z oczekiwaniami: gromadził się przy blokadzie proteasomów i szybko znikał po zatrzymaniu syntezy. APOL1 na powierzchni jednak prawie nie reagował na żaden z tych zabiegów. Gdy tylko cząsteczki APOL1 docierały do błony plazmatycznej, okazywały się wysoce odporne na szybki rozkład. Co więcej, chociaż warianty ryzyka wytwarzały mniej całkowitego APOL1 niż wersja normalna, ich poziomy powierzchniowe były podobne. Sugeruje to, że warianty ryzyka i normalne APOL1 są usuwane w podobnym tempie wewnątrz komórki, ale pule osadzone w błonie — które uważa się za formujące kanały jonowe i napędzające toksyczność — są zachowywane we wszystkich wariantach.
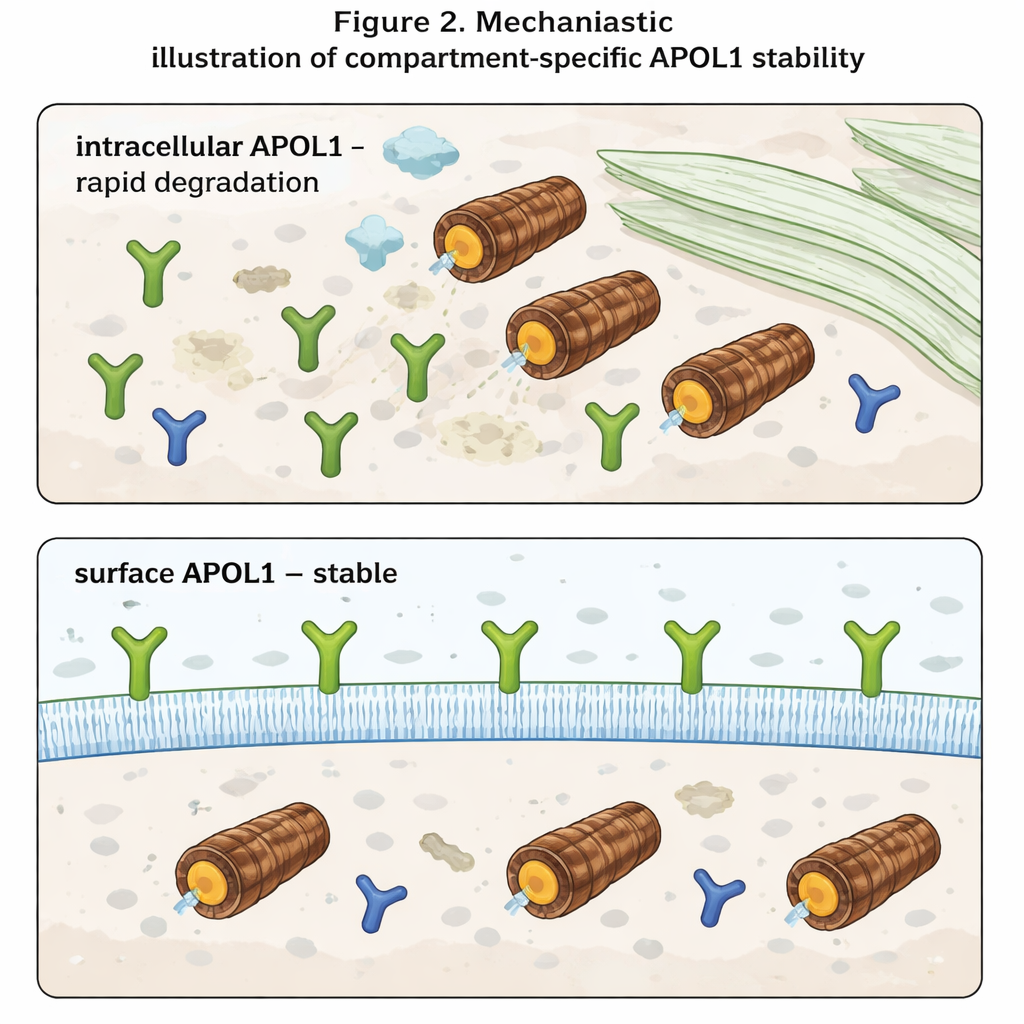
Co to oznacza dla przyszłych terapii
Dla odbiorców niebędących specjalistami kluczowy wniosek jest taki, że APOL1 zachowuje się zupełnie inaczej w zależności od miejsca występowania. Wewnątrz komórki jest białkiem krótkotrwałym, szybko rozpoznawanym i niszczonym. Na powierzchni komórki staje się długotrwałe i względnie chronione, nawet gdy zmienia się aparat degradacji komórkowej. Ponieważ choroba wydaje się powstawać, gdy kanały APOL1 na powierzchni zaburzają równowagę jonów, takich jak sód i potas, terapie mogą wymagać mniejszego skupienia na całkowitych poziomach APOL1, a większego na tym, ile z niego dociera do błony plazmatycznej i jak długo tam pozostaje. Strategie ograniczające transport APOL1 na powierzchnię lub selektywnie destabilizujące pulę powierzchniową mogłyby potencjalnie złagodzić uszkodzenia nerek bez całkowitego blokowania korzystnych funkcji tego genu w układzie odpornościowym.
Cytowanie: Höffken, V., Alvermann, L., Niggemeier, D. et al. APOL1 plasma membrane pools resist rapid protein degradation. Sci Rep 16, 6718 (2026). https://doi.org/10.1038/s41598-026-37647-z
Słowa kluczowe: APOL1, choroby nerek, rozpad białka, błona komórkowa, proteasom