Clear Sky Science · pl
Adaptatywne względem struktury genomu ramy do estymacji inbreedingu oparte na ROH u Penaeus vannamei
Dlaczego drzewa genealogiczne krewetek mają znaczenie dla twojego talerza
Nowoczesne farmy krewetek dostarczają znaczną część światowych owoców morza, ale powtarzalne rozmnażanie tych samych linii rodzinnych może cicho osłabiać ich kondycję. Gdy krzyżują się blisko spokrewnione osobniki, mogą zestawić się szkodliwe ukryte allele, co zmniejsza wzrost, przeżywalność i odporność na choroby. To badanie zadaje pozornie proste pytanie o duże znaczenie dla światowej akwakultury: jak mierzyć inbreeding u krewetek wystarczająco dokładnie, by utrzymać stada zdrowe i produktywne?
Ukryte genetyczne ślady inbreedingu
Inbreeding zostawia charakterystyczne odciski w DNA. Zamiast mieć dwie nieco różniące się wersje wielu genów, osobniki inbredowe mają tendencję do długich odcinków, gdzie obie kopie są identyczne. Genetycy nazywają te odcinki „runs of homozygosity” (ROH). Sumując, jaka część genomu osobnika przypada na takie odcinki, badacze mogą oszacować poziom inbreedingu bardziej precyzyjnie niż na podstawie papierowych rodowodów, które często są niekompletne lub zawierają błędy. Miara oparta na ROH, znana jako FROH, stała się standardem u bydła, świń i innych zwierząt hodowlanych, ale genomy krewetek stawiają szczególne wyzwania, które sprawiają, że gotowe metody bywają zawodnościowe.
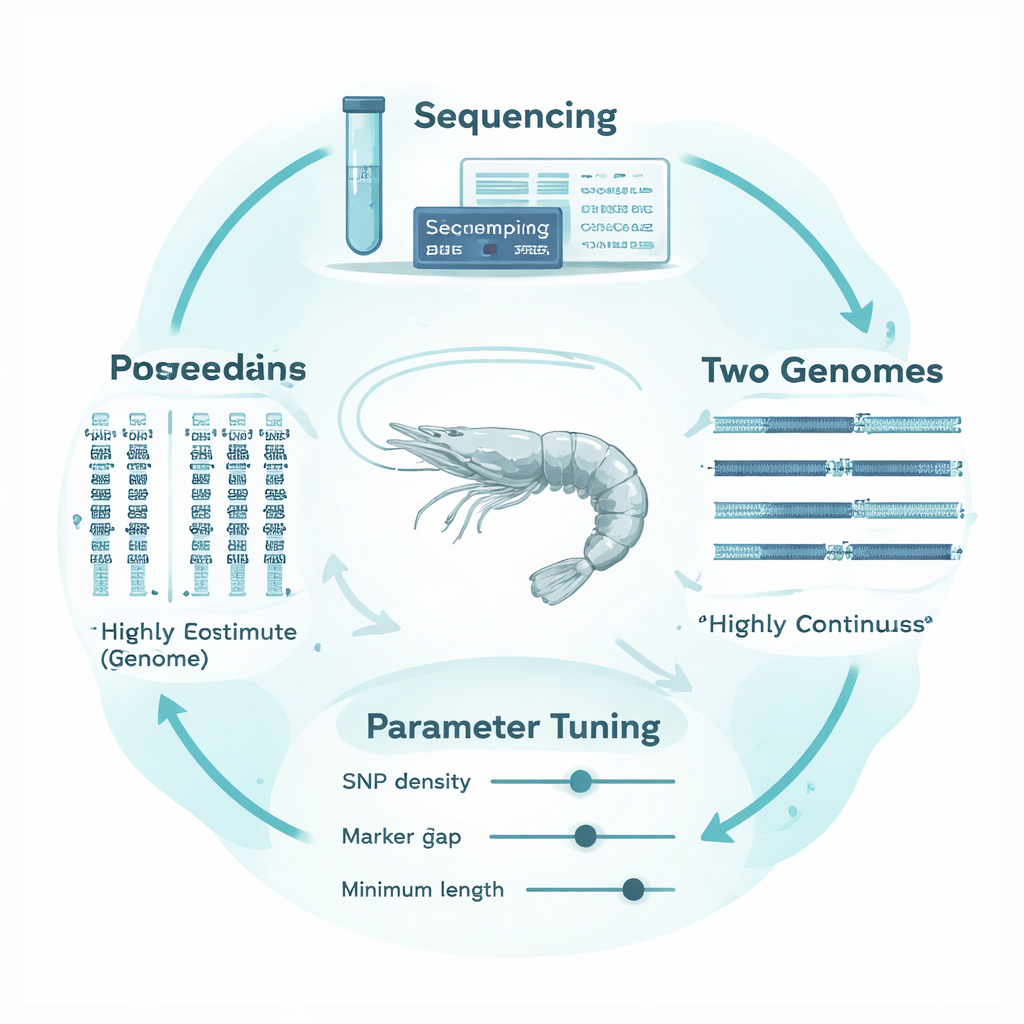
Dlaczego genomy krewetek są szczególnie problematyczne
Krewetka biała (Penaeus vannamei), najbardziej masowo hodowany gatunek krewetek na świecie, ma silnie fragmentaryczny i złożony genom. Zamiast długich, ciągłych sekwencji chromosomowych, wiele dostępnych map genomu jest rozbitych na tysiące mniejszych fragmentów, oddzielonych lukami i obszarami powtarzalnymi. Markery genetyczne są rozrzucone nierównomiernie po tym patchworku, a gatunek cechuje bardzo wysoka różnorodność genetyczna. Metody i ustawienia oprogramowania pierwotnie dostrojone do uporządkowanych, dobrze zmapowanych genomów ssaków mogą przez to mylnie traktować techniczne przerwy jako rzeczywiste końce ROH albo mylić krótkie, tło podobieństwa z prawdziwymi oznakami inbreedingu. W rezultacie istnieje wysokie ryzyko błędnej oceny stopnia inbreedingu u krewetki.
Budowanie miarki uwzględniającej strukturę genomu
Aby rozwiązać ten problem, autorzy zaprojektowali „adaptatywne względem struktury genomu” podejście, które dostosowuje analizę ROH do osobliwości DNA krewetek. Utworzyli trzynaście wysoce inbredowanych rodzin krewetek w kontrolowanych kojarzeniach, a następnie głęboko zsekwencjonowali genomy pięciu z tych rodzin oraz ich rodziców. Co istotne, te same dane sekwencyjne wyrównali do dwóch bardzo różnych genomów referencyjnych: jednego starszego, fragmentarycznego złożenia i nowszej, silnie ciągłej wersji. Korzystając z popularnego narzędzia PLINK, systematycznie testowali, jak osiem kluczowych ustawień wpływa na wykrywanie ROH, koncentrując się na trzech, które były szczególnie wrażliwe na strukturę genomu: jak gęsto muszą być rozmieszczone markery, jak dużą przerwę między markerami dopuszcza się wewnątrz runu i jak długi musi być run, by został policzony. Zbudowali empiryczne, niepokrywające się okna genomowe do śledzenia lokalnego rozmieszczenia markerów i braków danych oraz użyli „pokrycia genomowego” tych okien, wraz ze stabilnością FROH i długości ROH, jako obiektywnych wskazówek przy wyborze sensownych progów.
Różne ustawienia, ten sam obraz inbreedingu
Okazało się, że zoptymalizowane ustawienia były bardzo różne dla dwóch genomów referencyjnych. Genom fragmentaryczny wymagał znacznie większej gęstości markerów, krótszych dopuszczalnych przerw i krótszych minimalnych długości runów niż wysokiej jakości genom, aby uniknąć rozbijania prawdziwych ROH na wiele małych kawałków. Mimo to, po osobnym dostrojeniu tych parametrów, estymaty inbreedingu z obu referencji zbliżyły się do siebie: średnie FROH w inbredowanych krewetkach wynosiło około 0,24 w obu przypadkach, co ściśle odpowiadało oczekiwanej wartości z zaplanowanych kojarzeń i było ze sobą silnie zgodne. Jednocześnie ciągły genom ujawnił mniej, ale dużo dłuższych segmentów ROH, podczas gdy mapa fragmentaryczna pocięła wiele z nich na krótkie fragmenty. Badanie ujawniło też uderzające różnice wśród pełnych rodzeństw krewetek: nawet w ramach tej samej rodziny poziomy inbreedingu bardzo się różniły, czego proste zapisy rodowodowe nie potrafią uchwycić.
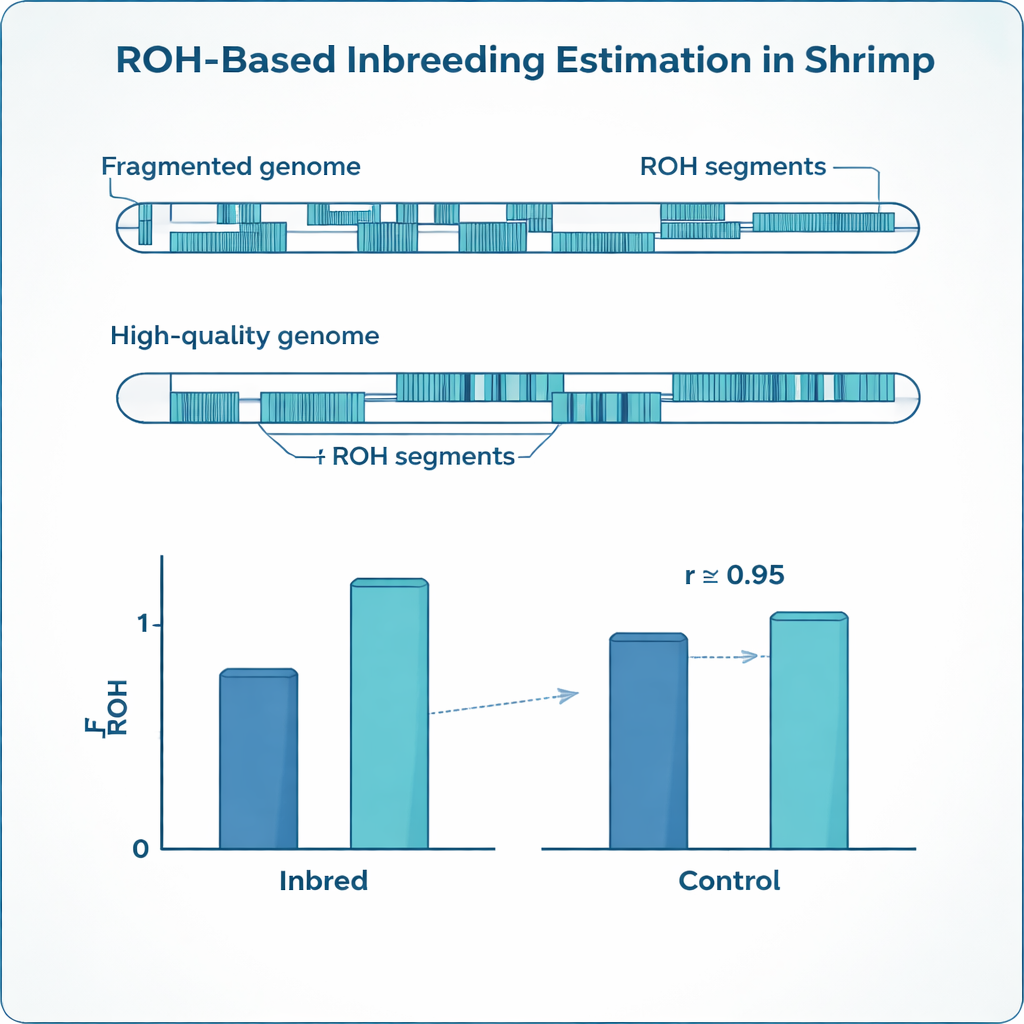
Bardziej precyzyjne narzędzia dla zdrowszych stad krewetek
Dla osób niebędących specjalistami najważniejszy wniosek jest prosty: mierzenie inbreedingu na podstawie DNA może być wysoce dokładne u krewetek hodowlanych, ale tylko wtedy, gdy metoda uwzględnia strukturę leżącego u podstaw genomu. Dostarczając praktycznej recepty na dostrojenie analizy ROH do fragmentarycznych genomów skorupiaków, praca ta pozwala hodowcom monitorować inbreeding osobnik po osobniku, zamiast polegać na niedoskonałych wykresach rodzinnych. To z kolei może pomóc zaprojektować plany kojarzeń, które utrzymują różnorodność genetyczną, jednocześnie poprawiając wzrost i odporność, wspierając bardziej zrównoważoną hodowlę krewetek i oferując wzorzec dla innych gatunków akwakultury borykających się z podobnymi problemami genomowymi.
Cytowanie: Zou, X., Zhou, H., Liu, M. et al. A genome-structure adaptive framework for ROH-based inbreeding estimation in Penaeus vannamei. Sci Rep 16, 6769 (2026). https://doi.org/10.1038/s41598-026-37622-8
Słowa kluczowe: hodowla krewetek, inbreeding, selekcja genomowa, runs of homozygosity, genetyka akwakultury