Clear Sky Science · pl
Ramowy model głębokiego uczenia oparty na DNABERT do przewidywania miejsc wiązania czynników transkrypcyjnych
Dlaczego przewidywanie przełączników kontroli DNA ma znaczenie
W każdej komórce twojego ciała znajduje się zasadniczo ten sam DNA, a mimo to komórki mózgu, wątroby i układu odpornościowego zachowują się zupełnie inaczej. Jednym z powodów są specjalne białka zwane czynnikami transkrypcyjnymi, które działają jak molekularne przełączniki—włączają lub wyłączają geny, dokując do krótkich odcinków DNA znanych jako miejsca wiązania. Eksperymentalne wykrycie wszystkich tych miejsc w całym genomie jest czasochłonne i kosztowne. W tym badaniu przedstawiono TFBS-Finder, nowy model sztucznej inteligencji, który potrafi czytać surowe litery DNA i dokładniej przewidywać, gdzie wiążą się czynniki transkrypcyjne, co może przyspieszyć badania nad regulacją genów i chorobami.
Czytanie DNA jak języka
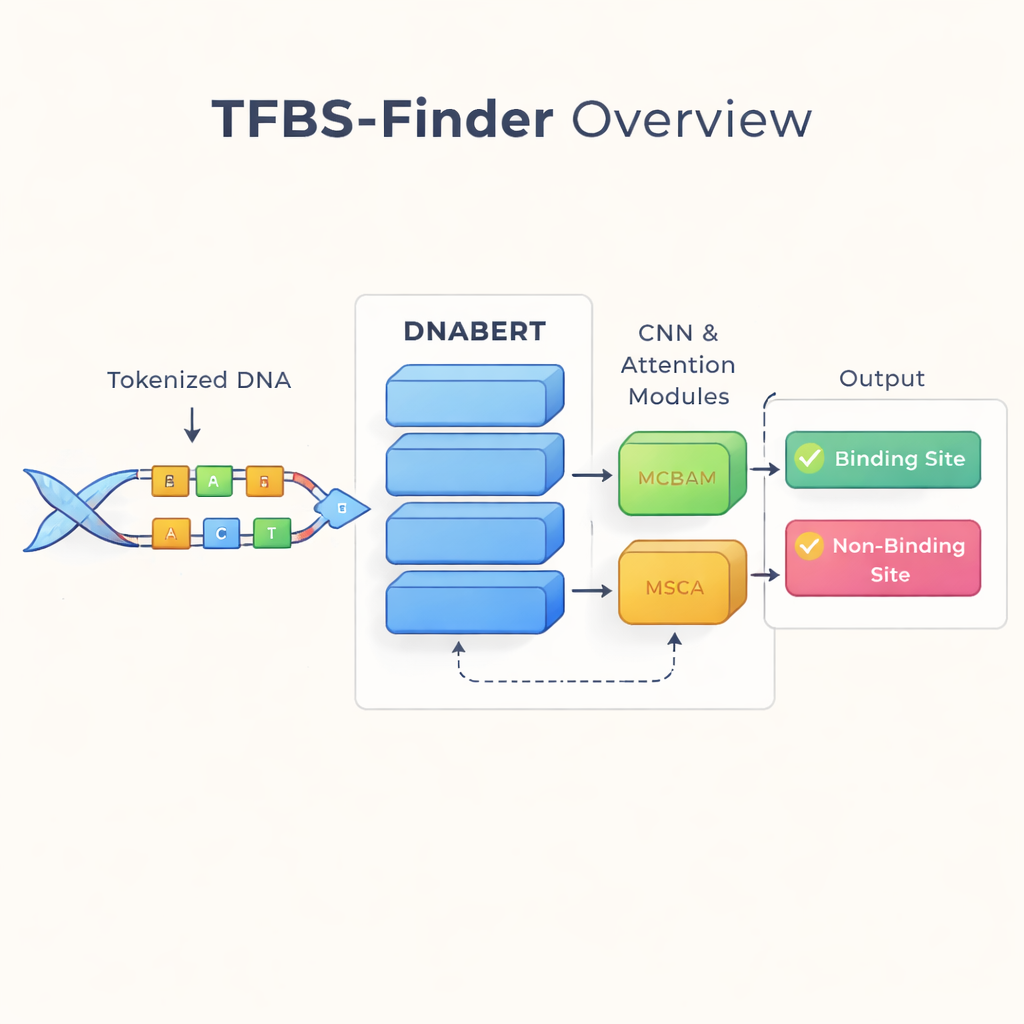
Autorzy bazują na pomyśle, który zrewolucjonizował technologie językowe: traktować DNA, jakby było tekstem. Wykorzystują DNABERT, wersję modelu BERT przetrenowaną na ludzkim DNA zamiast słów. DNABERT nie analizuje pojedynczych liter w izolacji; dzieli DNA na nakładające się krótkie „słówka” złożone z pięciu liter i uczy się, jak te fragmenty współwystępują. Pozwala to modelowi uchwycić długozasięgowy kontekst, na przykład jak wzorce na jednym końcu sekwencji odnoszą się do wzorców daleko, podobnie jak zrozumienie sensu zdania, a nie pojedynczych słów.
Wykrywanie lokalnych wzorców za pomocą ukierunkowanej uwagi
Choć DNABERT dobrze rozumie kontekst globalny, wiązanie czynników transkrypcyjnych często zależy od bardzo krótkich, precyzyjnych motywów—lokalnych wzorców w DNA. TFBS-Finder dodaje więc kilka dodatkowych komponentów na bazie DNABERT. Splotowa sieć neuronowa (CNN) przeszukuje osadzenia sekwencji, aby wyeksponować powtarzające się lokalne kształty, podobnie jak oprogramowanie do analizy obrazu wykrywa krawędzie i narożniki. Dwa moduły uwagi, nazwane MCBAM i MSCA, działają jak regulowane reflektory, wzmacniając najbardziej informacyjne cechy i tłumiąc szum. Razem te bloki równoważą szerokokontekstowe rozumienie z drobiazgowymi detalami, aby zdecydować, czy fragment DNA zawiera prawdziwe miejsce wiązania.
Udowadnianie, że każdy element naprawdę pomaga
Aby sprawdzić, czy wszystkie te komponenty są konieczne, zespół przeprowadził obszerne eksperymenty „ablacyjne”, systematycznie usuwając lub przestawiając moduły i ponownie trenując system na 165 zestawach benchmarkowych obejmujących 29 czynników transkrypcyjnych w 32 typach komórek. Przy użyciu standardowych miar jakości predykcji pełny model TFBS-Finder konsekwentnie wypadał najlepiej. Prostsze wersje opierające się wyłącznie na DNABERT lub pozbawione jednego z modułów uwagi wyraźnie traciły na dokładności. Testy statystyczne potwierdziły, że spadki wydajności nie były przypadkowe, pokazując, że połączenie globalnego rozumienia sekwencji i starannie zaprojektowanej uwagi do lokalnych wzorców jest kluczowe.
Praca w różnych typach komórek i przewyższanie starszych narzędzi
Istotnym pytaniem jest, czy model wytrenowany w jednym kontekście biologicznym potrafi uogólnić się na inny. Autorzy skupili się na dobrze przebadanym czynniku transkrypcyjnym o nazwie CTCF i trenowali TFBS-Finder na danych z jednej linii komórkowej, a następnie testowali go na innych. We wszystkich kombinacjach model osiągał wysokie wyniki, co sugeruje, że wychwytuje podstawowe cechy wiązania CTCF wspólne dla różnych tkanek. W porównaniu z dziewięcioma wiodącymi metodami, w tym wcześniejszymi modelami głębokiego uczenia i opartymi na BERT, TFBS-Finder wykazał wyższą średnią dokładność i generował bardziej wiarygodne rankingi miejsc wiązania. Działał też nieco szybciej i zużywał mniej pamięci niż najbardziej podobny poprzedni model, co wskazuje, że lepsza wydajność nie wymagała cięższego obliczeniowo podejścia.
Widzenie, czego model się nauczył
Złożone systemy AI bywają często krytykowane jako „czarne skrzynki”. Tutaj badacze próbowali je otworzyć, wizualizując, które pozycje w DNA najbardziej wpływały na decyzje TFBS-Finder. Dla dwóch czynników transkrypcyjnych o dobrze poznanych motywach wiązania, CEBPB i GATA3, wygenerowali miary ważności wzdłuż sekwencji i pogrupowali najsilniejsze sygnały w wzorce konsensusowe. Odtworzone motywy bardzo dobrze odpowiadały wzorcom referencyjnym z uznanych baz danych, a przewidywane regiony wiążące pokrywały się z niezależnie wykrytymi instancjami motywów. Sugeruje to, że TFBS-Finder nie tylko zapamiętuje przykłady, lecz nauczył się biologicznie istotnych zasad rozpoznawania DNA przez czynniki transkrypcyjne.
Co to znaczy dla genetyki i medycyny
TFBS-Finder dostarcza dokładniejszego i bardziej interpretowalnego sposobu mapowania przełączników kontrolnych zakodowanych w naszym DNA. Wskazując, gdzie czynniki transkrypcyjne mogą się wiązać, może pomóc badaczom w odwzorowywaniu sieci regulacji genów, priorytetyzowaniu wariantów genetycznych, które mogą zaburzać kluczowe miejsca kontrolne, oraz w projektowaniu bardziej ukierunkowanych eksperymentów. Chociaż obecna praca używa przetasowanych sekwencji jako sztucznych negatywów i koncentruje się tylko na literach DNA, autorzy planują dodać informacje o kształcie i strukturze DNA oraz badać bardziej realistyczne tła sekwencji. W miarę poprawy tych modeli mogą stać się potężnymi narzędziami do zrozumienia, jak zmiany w niekodującym DNA wpływają na rozwój, ewolucję i ryzyko chorób.
Cytowanie: Dutta, P., Ghosh, N. & Santoni, D. A DNABERT based deep learning framework for predicting transcription factor binding sites. Sci Rep 16, 7018 (2026). https://doi.org/10.1038/s41598-026-37483-1
Słowa kluczowe: miejsca wiązania czynników transkrypcyjnych, głębokie uczenie, DNABERT, regulacja genów, genomika