Clear Sky Science · pl
Tri‑omika i uczenie maszynowe identyfikują prognostyczne biomarkery i sygnatury metaboliczne w sepsie
Dlaczego ma to znaczenie dla osób z ciężkimi infekcjami
Sepsa to zagrażająca życiu reakcja na zakażenie, która może nadmiernie pobudzić układ odpornościowy i doprowadzić do niewydolności narządów. Lekarze wiedzą, że wczesne rozpoznanie sepsy i dostosowanie leczenia do pacjenta ratuje życie, ale obecne badania krwi są narzędziami mało precyzyjnymi: często niewiele mówią o tym, kto wyzdrowieje, a kto jest w największym ryzyku. To badanie wykorzystuje silne połączenie trzech typów pomiarów molekularnych oraz nowoczesne uczenie maszynowe, aby poszukać bardziej precyzyjnych sygnałów ostrzegawczych we krwi pacjentów z sepsą.
Patrząc na krew trzema różnymi perspektywami
Zamiast skupiać się na jednym rodzaju cząsteczek, badacze jednocześnie profilowali tych samych pacjentów na trzy sposoby. Zmierzyli, które geny są włączone lub wyłączone (transkryptomika), które białka są rzeczywiście obecne i aktywne (proteomika) oraz jakie małe cząsteczki metaboliczne krążą we krwi (metabolomika). Pobierali krew od 21 pacjentów z sepsą i 10 zdrowych ochotników, a zaawansowane metody statystyczne pozwoliły zobaczyć, jak te trzy warstwy zmieniają się wspólnie w chorobie. To „tri‑omiczne” spojrzenie pomaga przezwyciężyć istotny problem: w sepsie aktywność genów i poziomy białek mogą się rozłączyć, więc analiza tylko jednej warstwy może wprowadzać w błąd.
Uczenie algorytmów rozpoznawania wzorców wysokiego ryzyka
Z tysięcy genów i białek zespół najpierw zastosował metodę sieciową, aby znaleźć grupy elementów, które poruszały się razem w sepsie. Następnie porównali te grupy z białkami, które wyraźnie różniły się między pacjentami a zdrowymi kontrolami, co doprowadziło do 32 silnych kandydatów. Aby jeszcze bardziej zawęzić listę, sięgnęli po uczenie maszynowe, stosując dwa komplementarne algorytmy, które odsiewały słabsze sygnały i zostawiały tylko najbardziej informatywne. Gdy sprawdzili, jak pozostałe geny odnoszą się do przeżywalności w dużym publicznym zbiorze danych o sepsie, wyłoniły się dwa geny: TPR i ERN1. Pacjenci z wyższym poziomem TPR mieli tendencję do dłuższego przeżycia, natomiast wyższe poziomy ERN1 wiązały się z gorszymi wynikami.
Łączenie komórek odpornościowych z zaburzeniami metabolizmu
Badanie nie ograniczyło się do genów i białek. Skanując tysiące metabolitów we krwi pacjentów, badacze znaleźli 136 małych cząsteczek, które silnie korelowały z TPR i ERN1. Wiele z nich należało do szlaków związanych z tłuszczami błon komórkowych i kwasami tłuszczowymi, które są kluczowe dla sygnalizacji komórek odpornościowych i rozprzestrzeniania stanu zapalnego. Równocześnie analiza pojedynczych komórek — badająca indywidualne komórki odpornościowe zamiast uśredniać ich sygnał — wykazała, że TPR i ERN1 są szczególnie aktywne w monocytach, makrofagach i komórkach NK (natural killer). Razem te wyniki sugerują, że oba markery leżą na skrzyżowaniu między komórkami walczącymi z infekcją a sposobem, w jaki komórki te wykorzystują i przekształcają tłuszcze oraz energię podczas sepsy.
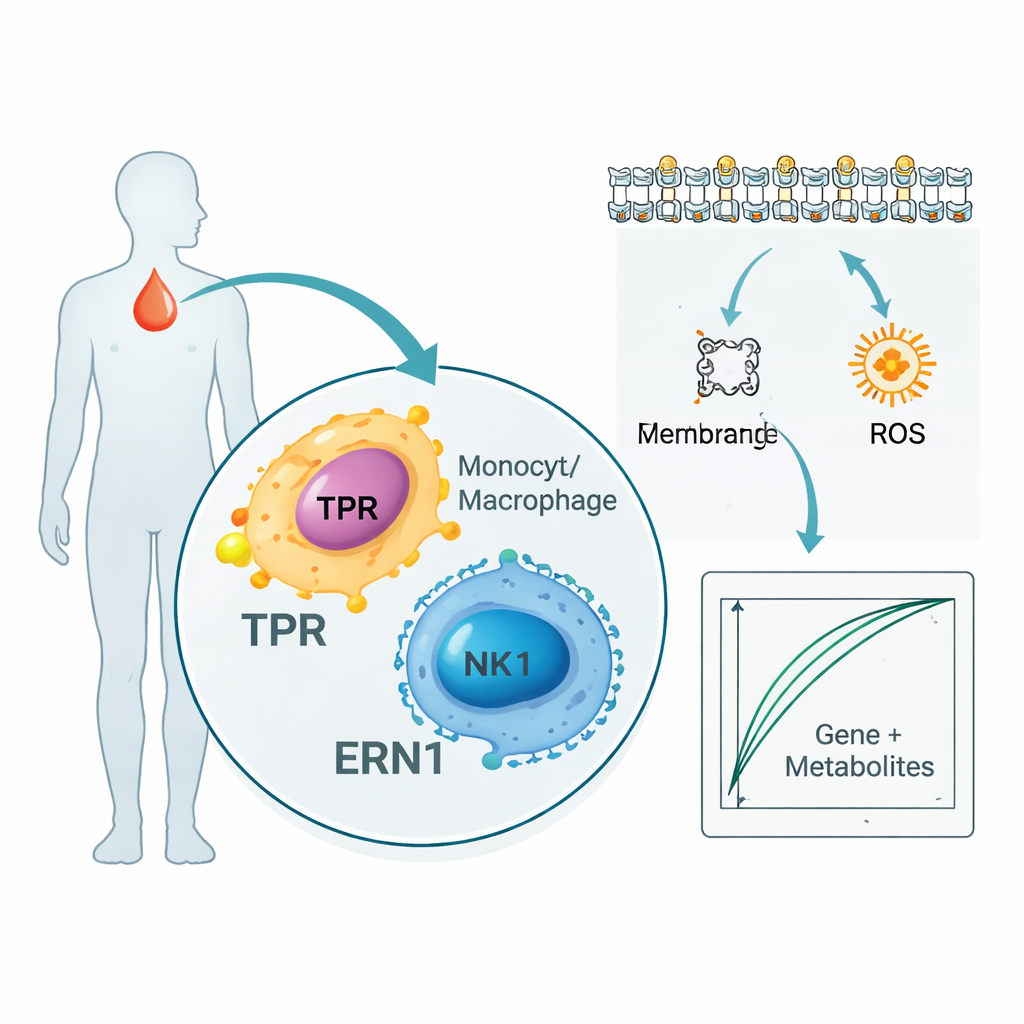
Budowa testu krwi jako dowodu koncepcji
Aby sprawdzić, jak odkrycia mogłyby przełożyć się na praktykę, autorzy połączyli dwa geny z pięcioma najbardziej informatywnymi metabolitami i wytrenowali proste modele komputerowe rozdzielające pacjentów z sepsą od zdrowych osób. W ich niewielkim wewnętrznym zbiorze danych te zintegrowane „gen‑plus‑metabolit” sygnatury potrafiły niemal doskonale rozróżnić, kto ma sepsę. Badacze sprawdzili też duże publiczne bazy, które łączą stężenia białek we krwi z ryzykiem chorób u dziesiątek tysięcy osób, i stwierdzili, że poziomy białek TPR i ERN1 były konsekwentnie powiązane ze stanami związanymi z sepsą, co daje dodatkowe wsparcie. Autorzy jednak podkreślają, że modele te są narzędziami wczesnej fazy, służącymi do generowania hipotez, a nie gotowymi testami przyłóżkowymi.
Związki z związkami roślinnymi jako wczesne tropy, nie kuracje
W ostatnim kroku zespół sprawdził, czy jakieś naturalne związki mogą wpływać na TPR lub ERN1. Przeszukali wyspecjalizowaną bazę niemal 500 oczyszczonych związków pochodzących z tradycyjnych medycyn chińskich, z których każdy miał profil aktywności genów. Kilka z tych związków wydawało się silnie podbijać lub tłumić oba geny w hodowanych komórkach, co sugeruje, że mogłyby kiedyś pomóc badaczom zgłębiać biologię sepsy lub projektować nowe leki. Jednak te wyniki pochodzą wyłącznie z dopasowań komputerowych: nie dowodzą, że którykolwiek z tych związków jest bezpieczny lub skuteczny u osób z sepsą.
Co naprawdę mówi nam ta praca
To badanie dostarcza raczej szczegółowej mapy niż gotowego rozwiązania. Poprzez splecenie trzech warstw molekularnych, danych z analizy pojedynczych komórek i uczenia maszynowego, autorzy wyróżniają TPR i ERN1 — oraz powiązane z nimi zmiany metaboliczne — jako obiecujące wskaźniki tego, jak układ odpornościowy i metabolizm ulegają rozregulowaniu w sepsie. Dla osoby niebędącej specjalistą kluczowy przekaz jest taki, że sepsa nie jest jednorodną jednostką chorobową, lecz zmiennym wzorcem stanów odpornościowych i metabolicznych, i że mądrzejsze testy krwi mogą w przyszłości pomóc lekarzom określić, w jakim stanie znajduje się pacjent, i odpowiednio dostosować leczenie. Zanim to nastąpi, te wstępne sygnały muszą zostać przetestowane i potwierdzone w znacznie większych, zróżnicowanych grupach pacjentów oraz w eksperymentach laboratoryjnych, które mogą wykazać związek przyczynowo‑skutkowy.
Cytowanie: Li, X., Ke, G., Hu, Y. et al. A tri-omics and machine learning framework identifies prognostic biomarkers and metabolic signatures in sepsis. Sci Rep 16, 6648 (2026). https://doi.org/10.1038/s41598-026-37342-z
Słowa kluczowe: markery sepsy, multi-omika, uczenie maszynowe w medycynie, metabolizm układu odpornościowego, diagnostyka precyzyjna