Clear Sky Science · pl
Adaptacyjna rozmyta klasteryzacja wspierająca prosty, szybki i wydajny wybór cech dla wysokowymiarowych i silnie niezrównoważonych dwuklasowych danych mikromacierzowych w bioinformatyce
Dlaczego to ma znaczenie dla badań nad genami
Nowoczesne testy ekspresji genów potrafią zmierzyć dziesiątki tysięcy genów w jednym próbce pacjenta. Ten potok danych obiecuje wcześniejszą diagnozę raka i lepszy dobór terapii, ale stwarza też problem: większość genów jest zaszumiona, nadmiarowa lub powiązana głównie z przypadkami powszechnymi, a nie z rzadkimi i groźnymi. W artykule przedstawiono nowy sposób przesiewania masywnych zbiorów danych ekspresji genów, tak aby komputery mogły wiarygodnie wykrywać pacjentów należących do małej, trudnej do wykrycia mniejszości, używając tylko niewielkiego, starannie wybranego zestawu genów.
Problem zbyt wielu, zbyt podobnych genów
W eksperymentach mikromacierzowych często monitoruje się tysiące poziomów aktywności genów dla zaledwie kilkuset pacjentów. Zwykle jedna klasa (np. powszechny podtyp nowotworu) znacznie przewyższa drugą, tworząc silnie niezrównoważone dane. W takim kontekście wiele genów zachowuje się bardzo podobnie, a wzorce dla pacjentów z klasy większości i mniejszości mogą się pokrywać. Standardowe metody uczenia mają tendencję do faworyzowania klasy większości i gubienia się w redundancji genów, co prowadzi do przeuczenia i słabej detekcji rzadkich podtypów. Tradycyjne metody redukcji wymiarów albo rezygnują z interpretowalności, tworząc nowe mieszane cechy, albo wybierają geny bez dokładnego sprawdzenia, jak pomagają klasyfikatorowi rozpoznać przypadki mniejszości.
Nowa mapa drogowa dla inteligentniejszego wyboru genów
Autorzy wprowadzają AFCG‑SFE — adaptacyjny model selekcji cech zaprojektowany specjalnie dla wysokowymiarowych, niezrównoważonych danych ekspresji genów. Metoda zaczyna od prostego „jednoagentowego” przeszukiwania, które włącza lub wyłącza geny i testuje, jak wspierają klasyfikację, ale wzbogaca to kilkoma krokami opartymi na danych. Najpierw grupuje geny według podobnego zachowania, a następnie pozwala genom należeć do więcej niż jednej grupy, aby odzwierciedlić biologiczną rzeczywistość, że gen może uczestniczyć w wielu ścieżkach. W obrębie każdej grupy klasyfikuje geny według tego, jak bardzo są informatywne względem etykiety choroby i zachowuje tylko kilku kluczowych reprezentantów, ostro redukując redundancję zanim rozpocznie się główne przeszukiwanie. 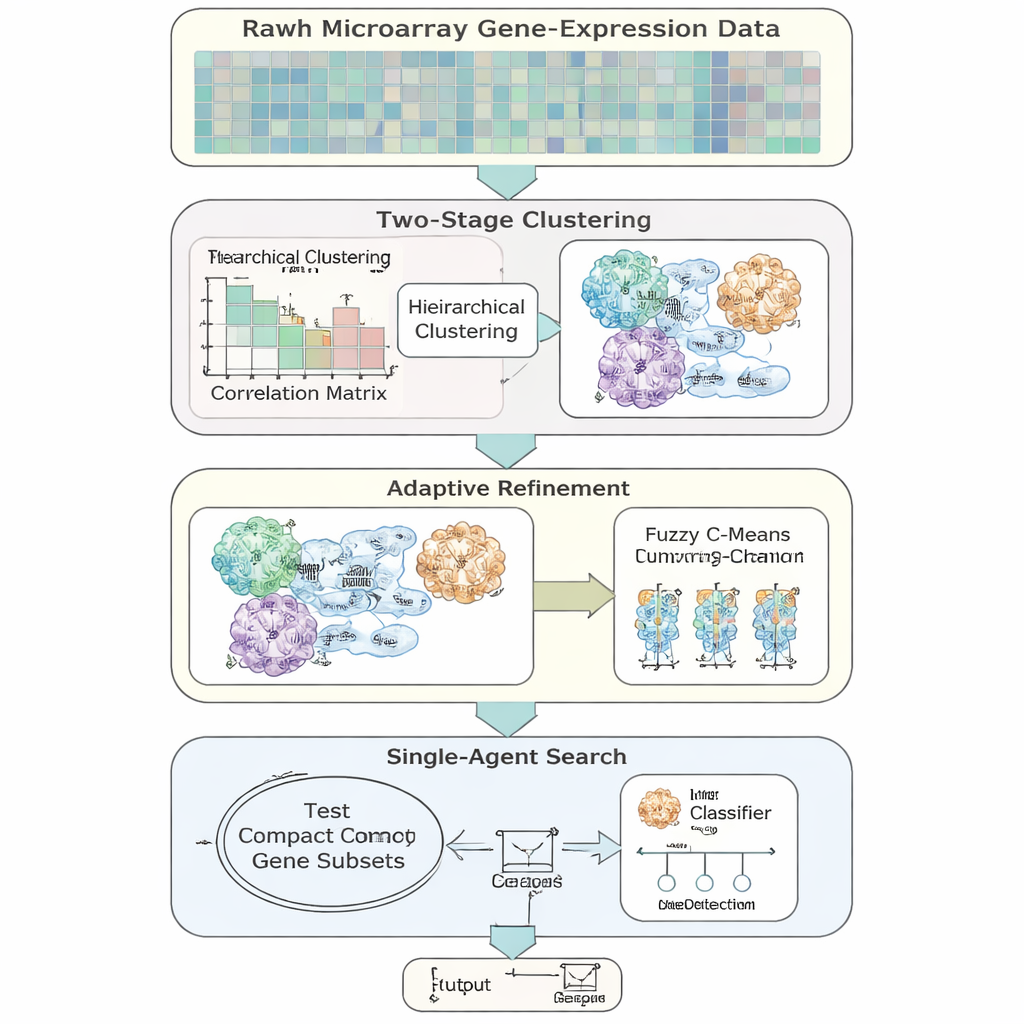
Sprawienie, by komputer zwracał uwagę na rzadkich pacjentów
Zamiast skupiać się na prostej dokładności, AFCG‑SFE używa funkcji dopasowania, która podkreśla miary odpowiednie dla nierównomiernych danych, w tym balans między poprawnym rozpoznaniem przypadków mniejszości i większości oraz wydajność dla wszystkich progów decyzyjnych. Funkcja dopasowania zawiera też kary za wybór zbyt wielu genów lub wielu genów z tego samego klastra oraz nagrodę za geny wykazujące silną zależność od etykiety choroby. Co ważne, siła tych kar i nagród jest ustawiana automatycznie na podstawie właściwości zbioru danych, takich jak liczba genów na pacjenta i stopień nakładania się klas, zamiast ręcznego strojenia. To sprawia, że metoda jest bardziej odporna i łatwiejsza do przeniesienia między badaniami.
Dopasowanie do trudności problemu
Kluczową ideą jest to, że algorytm nie powinien zawsze dążyć do najmniejszego możliwego zestawu genów. Gdy dwie klasy są bardzo trudne do rozdzielenia lub silnie się pokrywają, metoda automatycznie podnosi dolne ograniczenie liczby genów, które muszą zostać zachowane, zapewniając, że rzadkie, lecz istotne sygnały nie zostaną odrzucone. W miarę postępu przeszukiwania AFCG‑SFE stopniowo zaostrza per‑klastrowy limit liczby genów, które mogą przetrwać w każdej grupie, jednocześnie respektując ten minimalny próg. W rezultacie powstaje kompaktowy, różnorodny panel genów, który oddaje strukturę danych bez dominacji jednego, nadmiarowego wzorca. 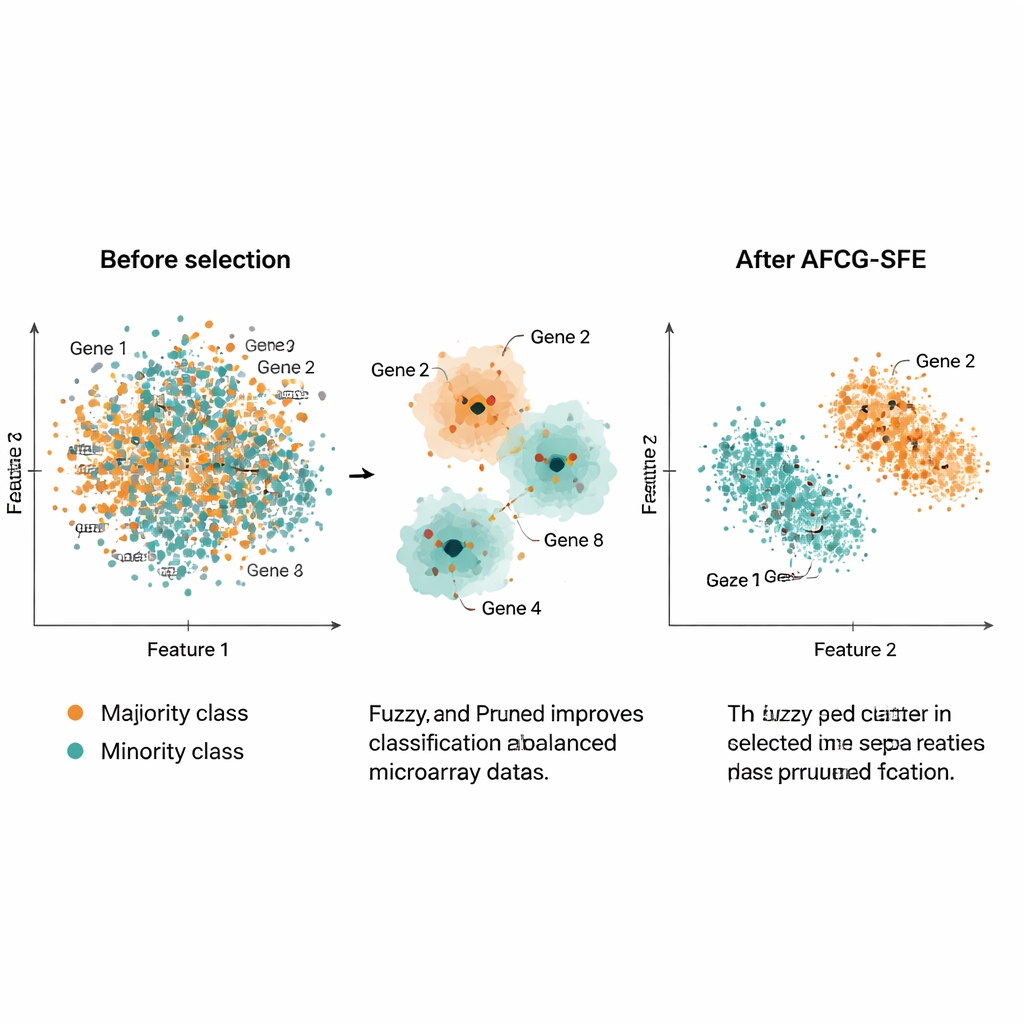
Co pokazują eksperymenty
Autorzy przetestowali AFCG‑SFE na 20 publicznych zestawach danych mikromacierzowych dotyczących nowotworów, z których każdy zawierał tysiące genów, ale tylko około 100–200 próbek i silne niezrównoważenie klas. Porównali swoją metodę z kilkoma ewolucyjnymi metodami przeszukiwania, prostymi filtrami i podejściami osadzonymi, które integrują selekcję cech z klasyfikatorem. W całym szeregu miar — w tym miara F, zbalansowana dokładność, pole pod krzywą ROC oraz miara przeuczenia — AFCG‑SFE był najlepszy lub współdzielił pierwsze miejsce we wszystkich zbiorach. Zazwyczaj wybierał mniej niż 25 genów (często zaledwie 6–8), eliminując ponad 99% pierwotnych cech, przy jednoczesnym poprawieniu lub utrzymaniu wydajności klasyfikacji. Zredukował też wskaźnik złożoności obrazujący, jak bardzo klasy pokrywają się w przestrzeni cech, co wskazuje na wyraźniejsze rozdzielenie po selekcji.
Wniosek dla osób niebędących ekspertami
W praktyce ta praca oferuje sposób na skompresowanie ogromnych, zaszumionych profili ekspresji genów do bardzo małych zestawów informatywnych genów, które nadal pozwalają komputerom dokładnie rozpoznawać rzadkie podgrupy pacjentów. Dzięki inteligentnemu grupowaniu podobnych genów, nagradzaniu tych, które rzeczywiście śledzą chorobę, oraz świadomej ochronie przed uprzedzeniem wobec klasy większości, AFCG‑SFE dostarcza zarówno lepszych prognoz, jak i znacznie prostszych paneli genów. Taka kombinacja może pomóc badaczom w identyfikacji potencjalnych biomarkerów, zaprojektowaniu bardziej interpretowalnych testów diagnostycznych i ostatecznie poprawie działania narzędzi medycyny precyzyjnej wobec rzeczywistych, niedoskonałych danych biologicznych.
Cytowanie: Tye, Y.W., Chew, X., Yusof, U.K. et al. Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data. Sci Rep 16, 6650 (2026). https://doi.org/10.1038/s41598-026-37086-w
Słowa kluczowe: ekspresja genów, selekcja cech, dane niezrównoważone, mikromacierze, podtypy nowotworów