Clear Sky Science · pl
rhinotypeR umożliwia powtarzalne przypisywanie genotypów rinowirusów na podstawie sekwencji VP4/2
Dlaczego nawet maleńkie wirusy przezięnień mają znaczenie
Większość z nas postrzega zwykłe przeziębienie jako uporczywą dolegliwość, a nie poważne zagrożenie. Tymczasem wirusy wywołujące wiele przeziębień — ludzkie rinowirusy — są też związane z ciężkimi zakażeniami płuc, napadami astmy i zaostrzeniami przewlekłych chorób płuc. Aby śledzić, jak te wirusy ewoluują i się rozprzestrzeniają, naukowcy muszą przypisywać im dokładne genetyczne „typy”, podobnie jak nadawanie kodów kreskowych produktom. Ten artykuł przedstawia rhinotypeR — darmowy, otwartoźródłowy pakiet oprogramowania, który sprawia, że takie genetyczne oznaczanie jest dokładniejsze, bardziej spójne i łatwiejsze do powtórzenia, pomagając zespołom zdrowia publicznego lepiej obserwować często pomijaną rodzinę wirusów oddechowych.
Ukryta różnorodność w zwykłych przeziębieniach
Ludzkie rinowirusy występują niezwykle często — wykrywa się je nawet w 60% próbek od osób z nagłymi dolegliwościami oddechowymi. Zamiast jednego wirusa, dzielą się na trzy główne grupy: A, B i C, oraz co najmniej 169 rozpoznanych typów genetycznych. Różne typy zachowują się inaczej: niektóre częściej wiążą się z ciężkimi zakażeniami u dzieci i z zaostrzeniami astmy, inne rzadziej pojawiają się przy poważnych chorobach. Ponieważ typy te ewoluują niezależnie i mają odrębne cechy powierzchniowe, naukowcy potrzebują wiarygodnych metod ich rozróżniania, aby śledzić, jak ogniska przemieszczają się w szkołach, gospodarstwach domowych i społecznościach.
Od rozproszonych narzędzi do jednej przejrzystej ścieżki
Dotychczas przypisywanie typu rinowirusa na podstawie kodu genetycznego było zadaniem złożonym i rozproszonym. Badacze zwykle koncentrowali się na krótkim fragmencie genomu wirusa zwanym regionem VP4/2, dopasowywali go do znanych szczepów referencyjnych, mierzyli różnice między sekwencjami, a następnie stosowali progi decyzyjne, by określić typ próbki. Te kroki były wykonywane przy użyciu mieszanki programów, ręcznych korekt i subiektywnej oceny. To utrudniało porównywanie i powtarzalność badań, nawet przy podobnych danych. rhinotypeR został stworzony, by przekształcić ten wieloetapowy, podatny na błędy proces w pojedynczy, skryptowalny workflow, który każdy może uruchomić i udostępnić.
Co właściwie robi nowe oprogramowanie
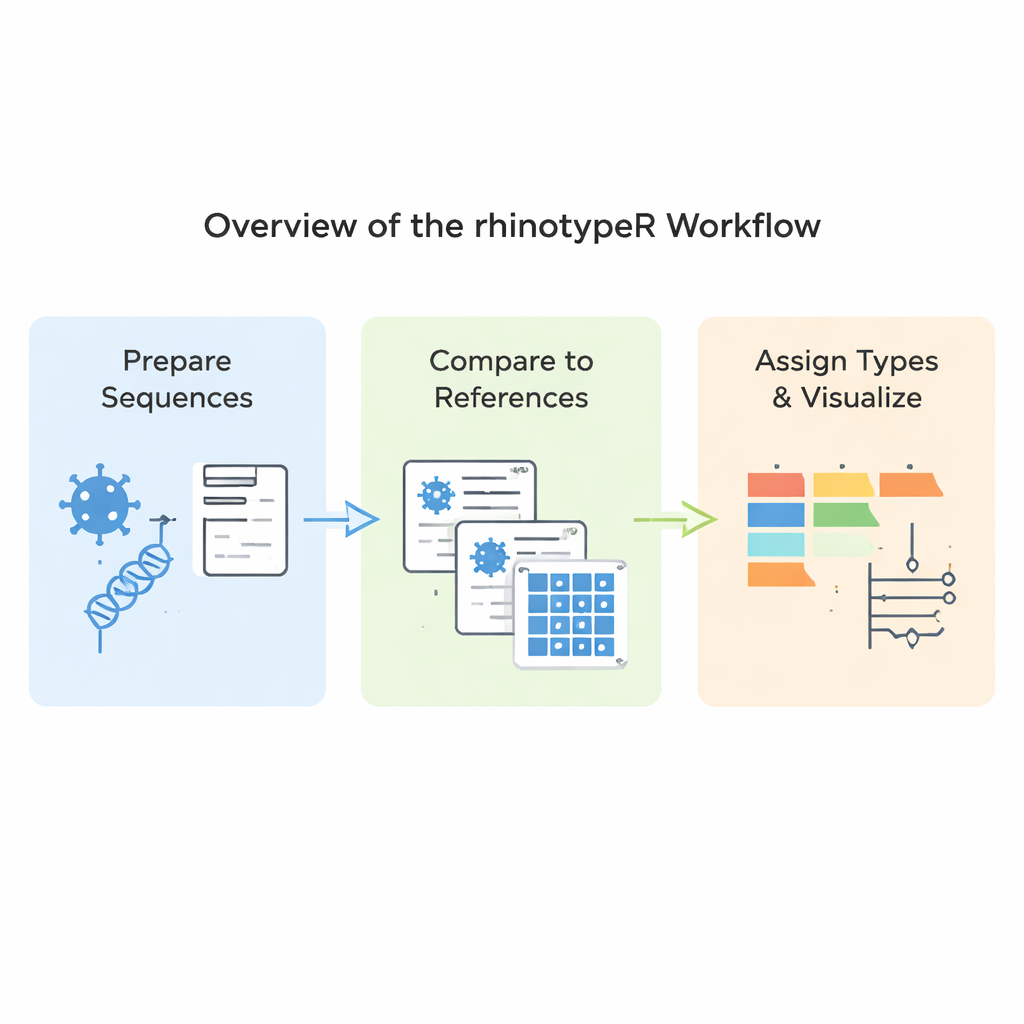
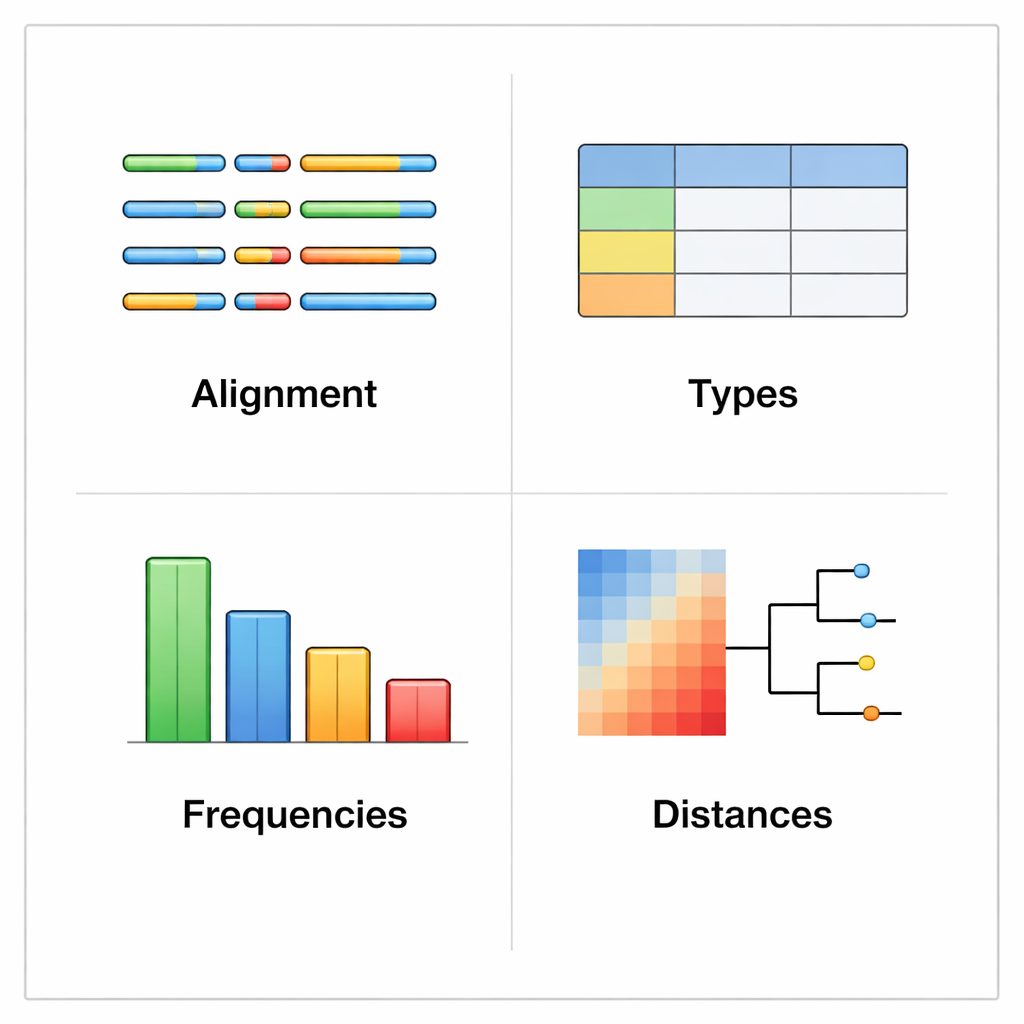
rhinotypeR działa w popularnym środowisku R i Bioconductor przeznaczonym do analiz danych. Przyjmuje kolekcję sekwencji VP4/2 rinowirusów i prowadzi je przez trzy główne etapy: przygotowanie i wyrównanie sekwencji, obliczenie odległości każdej sekwencji względem skuratorowanego zestawu referencyjnych typów oraz przypisanie każdej próbki do najbliższego znanego typu albo oznaczenie jej jako „nieprzypisana”, jeśli jest zbyt odmienna. To samo narzędzie potrafi wygenerować wyjścia wizualne, w tym kolorowe mapy różnic genetycznych, proste drzewa filogenetyczne i wykresy pokazujące częstość występowania poszczególnych typów w zbiorze danych. Użytkownicy mogą wyrównać swoje dane za pomocą zewnętrznych programów, jeśli wolą, albo pozwolić rhinotypeR przeprowadzić cały proces w R dla maksimum powtarzalności.
Testowanie narzędzia w praktyce
Aby sprawdzić wiarygodność wyników rhinotypeR, autorzy porównali jego pomiary odległości z wynikami z dwóch uznanych programów, ape i MEGA X, używając tych samych plików wejściowych i modeli. Wyniki były niemal identyczne; drobne rozbieżności wynikały ze zwykłego zaokrąglania w obliczeniach komputerowych, a nie z różnic metodycznych. Zespół uruchomił następnie rhinotypeR na dużym zbiorze ponad 2 300 sekwencji rinowirusów z wielu wcześniejszych badań, obejmującym ponad 90% znanych typów. W około czterech na pięć przypadków nowe narzędzie zgadzało się dokładnie z wcześniejszymi oznaczeniami typów. Większość niezgodności występowała tuż przy wcześniej ustalonych progach rozdzielenia typów, czyli tam, gdzie oczekiwane są decyzje graniczne. Co istotne, próbki, których nie można było pewnie przypisać do znanego typu, nie wykazywały oznak złej jakości czy niskiej zawartości wirusa, co sugeruje, że mogą odzwierciedlać rzeczywistą wirusową różnorodność.
Dlaczego to ważne dla zdrowia publicznego
Dla osób spoza specjalności kluczowa wiadomość jest taka, że rhinotypeR nie wymyśla na nowo sposobu klasyfikacji wirusów przeziębień; raczej czyni ten proces jaśniejszym, bardziej przejrzystym i łatwiejszym do powtórzenia. Łącząc wyrównanie, obliczenia odległości i przypisywanie typów w jednym otwartoźródłowym pakiecie — wraz z czytelnymi podsumowaniami wizualnymi — pomaga badaczom i programom nadzoru przetwarzać tysiące próbek w spójny sposób. Taka spójność polepsza naszą zdolność do porównywania badań z różnych miejsc i okresów, wczesnego wykrywania nietypowych lub nowych linii wirusów oraz łączenia wzorców genetycznych z rzeczywistymi trendami chorobowymi. W dłuższej perspektywie narzędzia takie jak rhinotypeR wzmacniają rutynowy monitoring pozornie zwykłych przeziębień, które u wielu osób mogą prowadzić do poważnych chorób.
Cytowanie: Luka, M.M., Nanjala, R., Rashed, W.M. et al. rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences. Sci Rep 16, 6149 (2026). https://doi.org/10.1038/s41598-026-37050-8
Słowa kluczowe: genotypowanie rinowirusów, nadzór molekularny, sekwencjonowanie VP4/2, narzędzia bioinformatyczne, wirusy układu oddechowego