Clear Sky Science · pl
Podejście uczenia maszynowego do przewidywania współczynników osmotycznych i wyprowadzania współczynników aktywności w solach amoniowych alkilowych
Codzienne chemikalia o ukrytej złożoności
Od zmiękczaczy tkanin i odżywek do włosów po chusteczki dezynfekujące i płyny do płukania ust — rodzina związków nazywana solami czwartorzędowych amoniowych, często skracana do „Quatów”, dyskretnie napędza wiele produktów, z których korzystamy. Pomagają zabijać zarazki, zmiękczać tkaniny i przyspieszać reakcje przemysłowe. Jednak przewidywanie, jak dokładnie te sole zachowują się w wodzie, okazało się zaskakująco trudne, co ogranicza efektywne projektowanie bezpieczniejszych i bardziej ekologicznych formulacji. W tym badaniu pokazano, że współczesne metody uczenia maszynowego potrafią uczyć się na podstawie wcześniejszych pomiarów, by przewidywać to zachowanie w sposób bardziej elastyczny i w wielu przypadkach dokładniejszy niż tradycyjne modele.
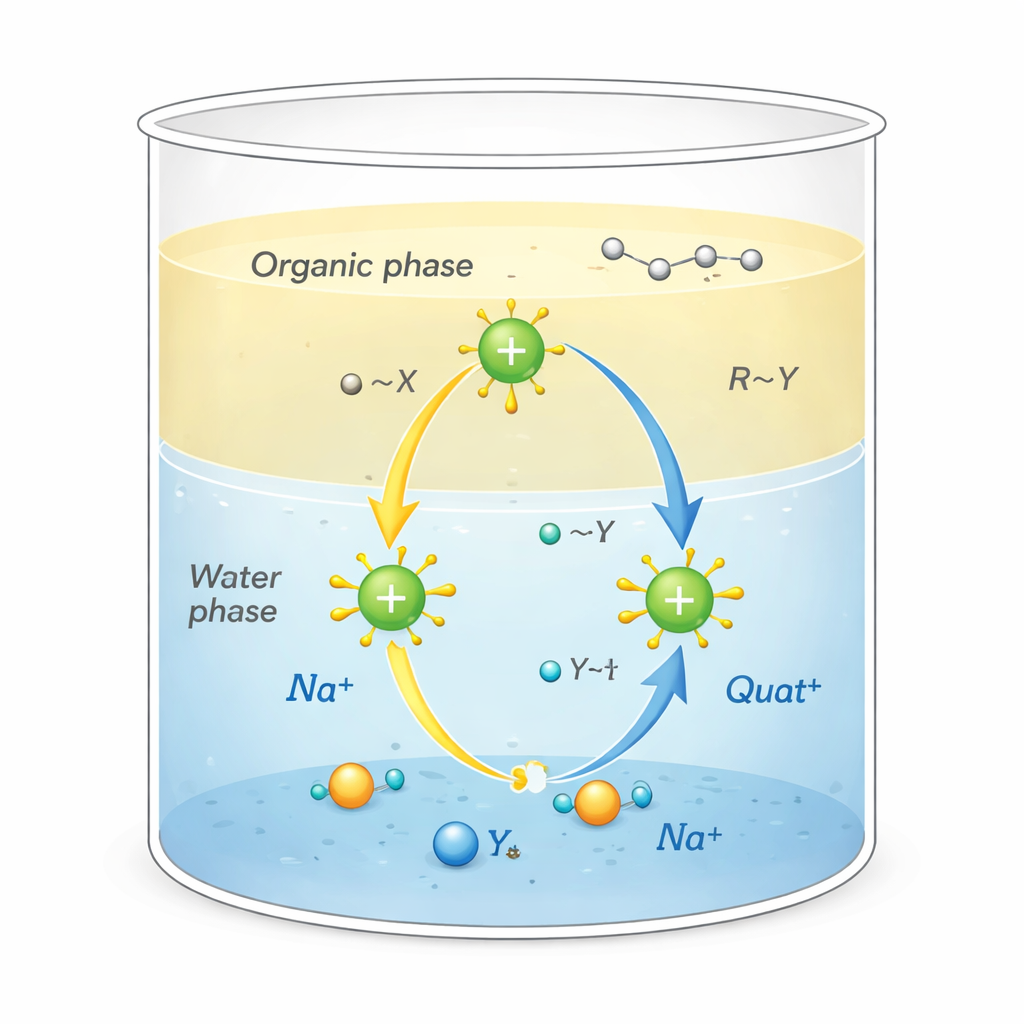
Dlaczego te sole są ważne
Quaty to dodatnio naładowane cząsteczki otoczone węglowodorowymi „ogonami”. Ta nietypowa budowa pozwala im pełnić kilka ról jednocześnie: przywierać do tłustych zabrudzeń, przylegać do powierzchni takich jak tkaniny czy włosy i zaburzać błony drobnoustrojów, dzięki czemu są skutecznymi środkami dezynfekującymi i powierzchniowo czynnymi. Są również stosowane jako katalizatory przeniesienia fazowego, działając jak kurierzy przenoszący reakcjonujące jony z wody do rozpuszczalników oleistych, do których normalnie nie przenikają. To przenoszenie, zachodzące na granicy wody i oleju, może znacząco przyspieszyć reakcje chemiczne wykorzystywane w produkcji farmaceutyków, polimerów i substancji chemicznych wysokiej czystości.
Dlaczego trudno przewidzieć ich zachowanie
Aby zaprojektować nowe Quaty lub dostroić istniejące, chemicy muszą wiedzieć, jak zachowują się one w roztworze — jak silnie oddziałują z wodą i z innymi rozpuszczonymi jonami. Dwa kluczowe wskaźniki to współczynnik osmotyczny, który odzwierciedla, jak sole wpływają na skłonność wody do przemieszczania przez błony, oraz współczynnik aktywności, który ujmuje, jak „efektywna” jest gatunek rozpuszczony w porównaniu z idealnym, doskonale wymieszanym roztworem. Tradycyjnie wartości te uzyskuje się albo przez mozolne pomiary eksperymentalne, albo za pomocą złożonych modeli fizycznych, takich jak Electrolyte‑NRTL i Extended UNIQUAC, które wymagają wielu dopasowanych parametrów i trudno je uogólnić na nowe cząsteczki.
Nauczanie komputera „czytania” cząsteczek
Badacze poszli inną drogą: zapytali, czy komputer może nauczyć się bezpośredniego związku między strukturą Quatów a ich zachowaniem osmotycznym na podstawie dostępnych danych. Zgromadzili 1 654 pomiarów współczynników osmotycznych dla 52 różnych Quatów z literatury naukowej. Każda cząsteczka została opisana notacją SMILES — tekstowym zapisem, który koduje cechy takie jak liczba atomów węgla i tlenu, obecność pierścieni benzenowych, rozgałęzienie oraz typ dodatnio naładowanej grupy azotowej, wraz z towarzyszącym anionem (np. chlorkowym, bromkowym czy azotanowym). Te deskryptory strukturalne, wraz ze stężeniem soli, posłużyły jako dane wejściowe do kilku nadzorowanych algorytmów uczenia maszynowego zaimplementowanych w Pythonie.
Poszukiwanie najbardziej niezawodnego predyktora
Przeszkolono siedem różnych algorytmów, w tym regresję liniową, drzewa decyzyjne, lasy losowe, maszyny wektorów nośnych, gradient boosting, k‑najbliższych sąsiadów oraz procesy Gaussowskie. Dane podzielono na 70% do treningu i 30% do testów. Zastosowano też bardziej rygorystyczny schemat walidacji, w którym wszystkie dane dla jednej soli były odrzucane podczas trenowania, aby sprawdzić, jak dobrze modele ekstrapolują do prawdziwie nieznanej substancji. Regresja liniowa wypadła słabo, nie wychwytując ważnych nieliniowych zależności. Metody oparte na drzewach bardzo dobrze dopasowały się do danych treningowych, ale dawały nieco poszarpane prognozy i traciły dokładność dla nowych soli. Model oparty na procesie Gaussowskim osiągnął najlepszy kompromis: dostarczał gładkie, fizycznie rozsądne krzywe współczynników osmotycznych i osiągnął średni bezwzględny błąd procentowy rzędu 5% ogółem, przewyższając alternatywne podejścia uczenia maszynowego w najtrudniejszych testach.
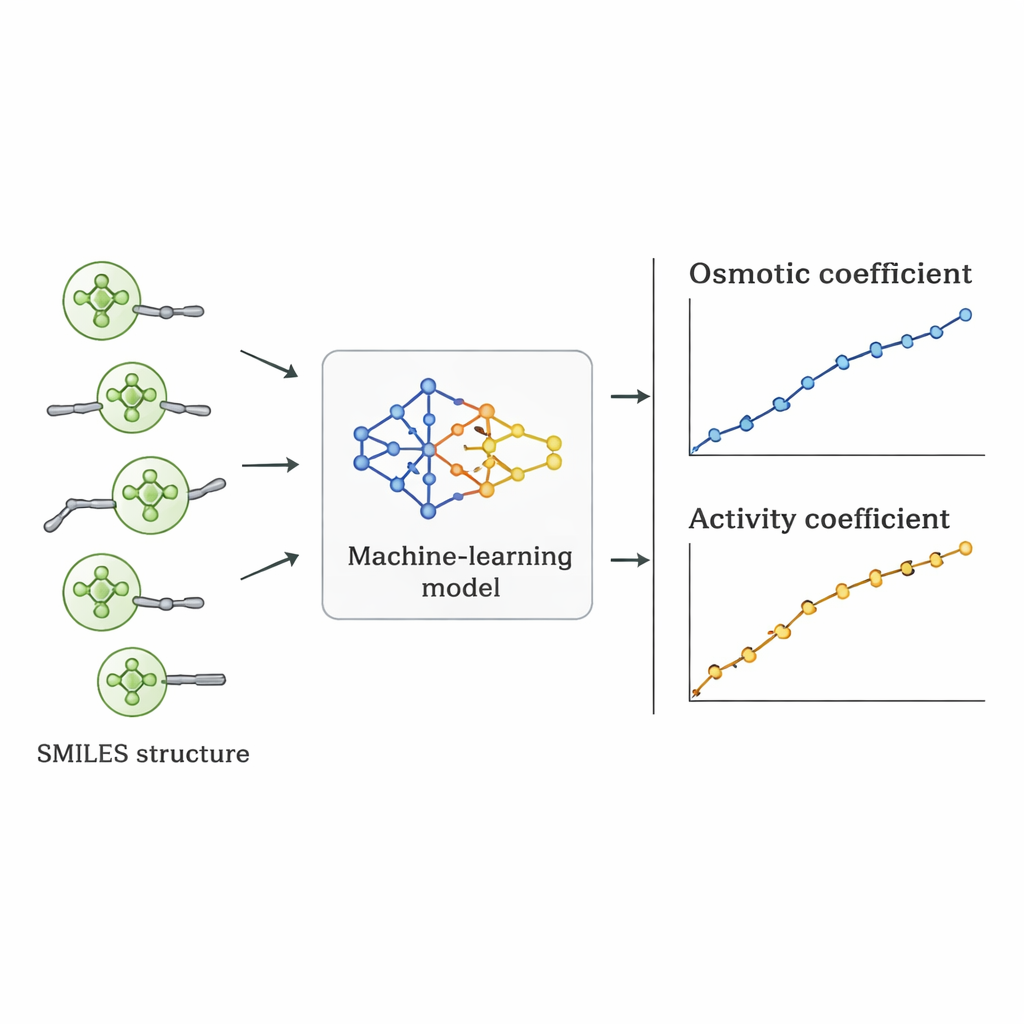
Od zachowania osmotycznego do użytecznych liczb projektowych
Po wybraniu najlepszego modelu jego przewidywane współczynniki osmotyczne przekształcono w współczynniki aktywności, korzystając ze standardowych zależności termodynamicznych. Porównanie tych współczynników aktywności z wartościami uzyskanymi eksperymentalnie i z ustalonych modeli fizycznych wykazało, że podejście oparte na uczeniu maszynowym często dorównywało im lub je przewyższało dla poszczególnych Quatów. Chociaż jego średni błąd dla wszystkich substancji był nieco większy niż w przypadku niektórych wyspecjalizowanych modeli, miał istotną zaletę: ponieważ opiera się na deskryptorach strukturalnych, a nie na dopasowaniu specyficznym dla danej soli, można go stosować do nowych Quatów, które nigdy nie były mierzone w laboratorium, pod warunkiem że ich struktury przypominają te z zestawu treningowego.
Co to oznacza dla produktów i procesów
Dla osoby niebędącej specjalistą główny wniosek jest taki, że komputery potrafią teraz „czytać” zwarte tekstowe opisy cząsteczek i na podstawie wzorców wyuczonych z wcześniejszych danych przewidywać, jak te cząsteczki zachowają się w wodzie, z imponującą dokładnością. To otwiera drogę do szybszego i tańszego przesiewania nowych Quatów do środków dezynfekujących, detergentów, produktów pielęgnacji osobistej i katalizatorów przemysłowych, bez konieczności prowadzenia wyczerpujących eksperymentów dla każdego kandydata. Obecny model to tylko pierwszy krok, a autorzy zauważają, że bogatsze odciski molekularne i nowsze algorytmy mogą jeszcze poprawić wydajność. Mimo to pokazuje, jak narzędzia oparte na danych mogą uzupełniać tradycyjną chemię, pomagając inżynierom projektować bardziej efektywne i potencjalnie bezpieczniejsze formulacje, eksplorując możliwości chemiczne, które byłyby niepraktyczne do testowania pojedynczo w laboratorium.
Cytowanie: Chawuthai, R., Murathathunyaluk, S., Saengsuradech, S. et al. A machine learning approach for predicting osmotic coefficients and deriving activity coefficients in alkyl ammonium salts. Sci Rep 16, 5969 (2026). https://doi.org/10.1038/s41598-026-36758-x
Słowa kluczowe: sole czwartorzędowych amoniowych, kataliza przeniesienia fazowego, współczynniki osmotyczne, współczynniki aktywności, uczenie maszynowe w chemii