Clear Sky Science · pl
Hybrydowe ramy głębokiego uczenia do dokładnej klasyfikacji wysokowymiarowych danych genomowych
Rozszyfrowywanie zalewu danych genomowych
Współczesne technologie DNA potrafią zmierzyć dziesiątki tysięcy genów w jednym eksperymencie, co otwiera perspektywy wcześniejszego wykrywania chorób i bardziej precyzyjnego leczenia. Jednak ta obfitość danych jest tak duża, zaszumiona i złożona, że nawet potężne modele komputerowe często mają trudności ze znalezieniem wyraźnych, godnych zaufania wzorców. W artykule przedstawiono nowy rodzaj systemu sztucznej inteligencji zaprojektowany specjalnie do przetwarzania tak przytłaczających danych genomowych, z celem polepszenia trafności prognoz i jednoczesnego wyjaśniania, jak te prognozy powstały.
Dlaczego dane genomowe są trudne w użyciu
Badania genomowe rutynowo generują znacznie więcej pomiarów niż jest pacjentów lub próbek. Wiele z tych pomiarów jest nieistotnych, redundantnych lub zniekształconych przez szumy techniczne. Tradycyjne metody uczenia maszynowego albo wymagają, by eksperci ręcznie wskazywali, które geny mogą być istotne, albo próbują wykorzystać wszystko naraz i ryzykują nadmierne dopasowanie — czyli dobre wyniki na danych treningowych, a złe na nowych przypadkach. Głębokie uczenie, które zrewolucjonizowało dziedziny takie jak rozpoznawanie obrazów, potrafi automatycznie wydobywać wzorce z surowych danych. Jednak w genomice często zachowuje się jak czarna skrzynka: może dawać dokładne odpowiedzi, ale niewiele wyjaśniać dlaczego, co ogranicza jego akceptację w medycynie, gdzie przejrzystość jest kluczowa.
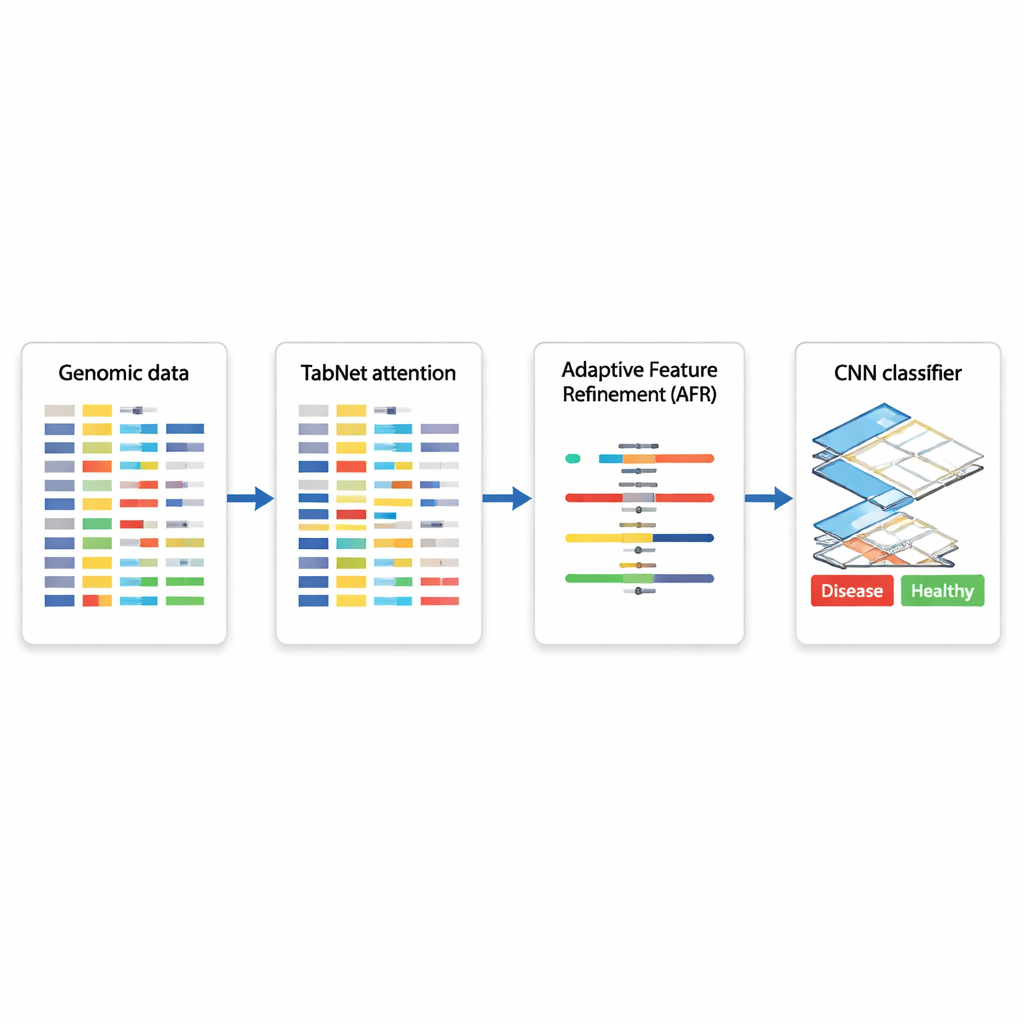
Hybrydowy plan AI do decyzji opartych na genach
Autorzy proponują hybrydową architekturę głębokiego uczenia łączącą trzy wyspecjalizowane moduły. Najpierw komponent o nazwie TabNet działa jak reflektor, skanując wszystkie dostępne pomiary genomowe i ucząc się, które cechy są najbardziej informatywne dla danego zadania — na przykład rozróżnienia tkanki nowotworowej od nienowotworowej. Zamiast traktować każdy gen jednakowo, TabNet koncentruje uwagę na rzadkiej podzbiorze cech, które wydają się najbardziej istotne. Następnie warstwa Adaptacyjnego Rafinowania Cech (AFR) przejmuje te wybrane sygnały i ponownie je ważaruje, wzmacniając spójne, istotne wzorce, jednocześnie dalej tłumiąc szumy. Na końcu konwolucyjna sieć neuronowa (CNN), powszechnie stosowana w analizie obrazów, bada, jak udoskonalone cechy oddziałują lokalnie, wychwytując subtelne relacje między grupami genów, które mogą sygnalizować określony podtyp choroby lub stan biologiczny.
Testowanie modelu
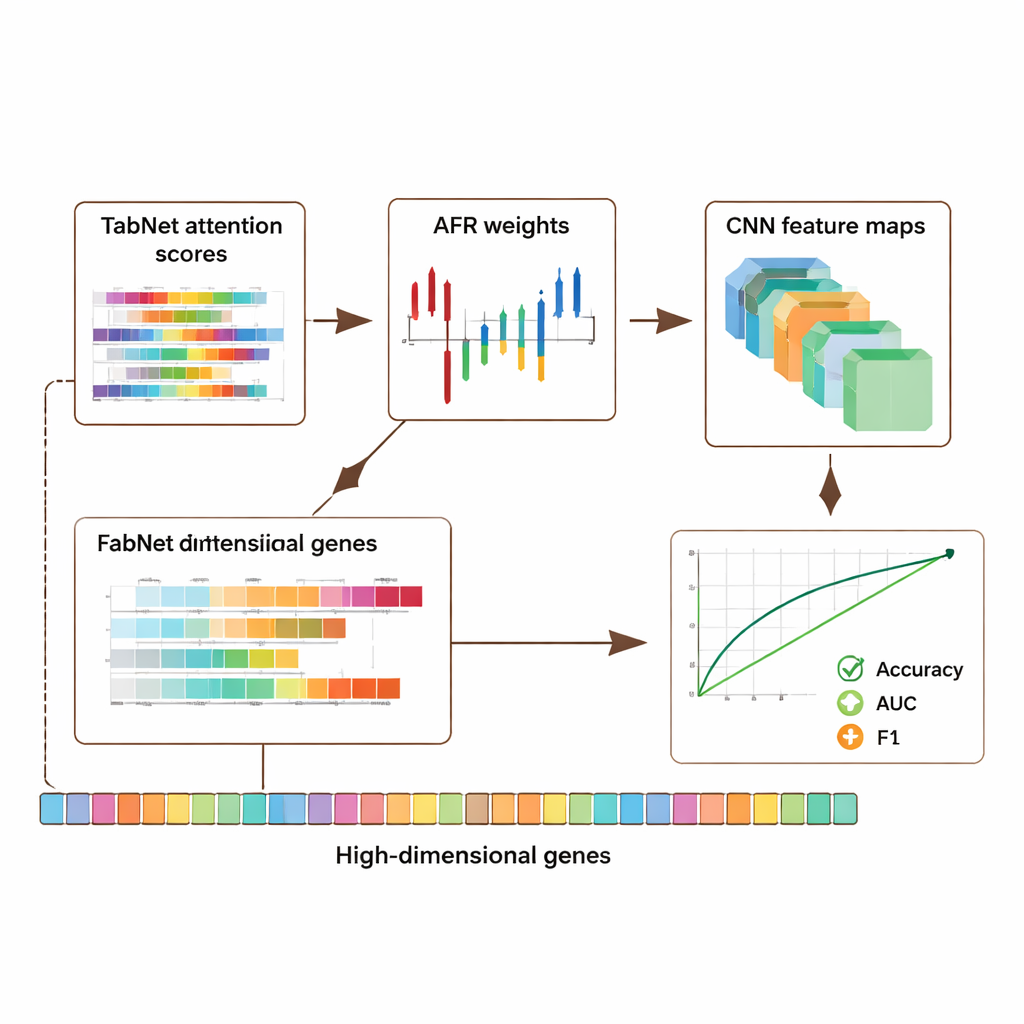
Ramę oceniono na trzech głównych publicznych zasobach: zestawie danych raka piersi z The Cancer Genome Atlas, zestawie komórek pojedynczych melanomu z Gene Expression Omnibus oraz zestawie epigenomicznym z projektu ENCODE. Razem te kolekcje obejmują tysiące próbek i dziesiątki tysięcy cech na próbkę, obejmując aktywność genów i chemiczne znaki na DNA. We wszystkich zestawach hybrydowy model przewyższał kilka najnowocześniejszych podejść, poprawiając dokładność i kluczowe miary jakości klasyfikacji, takie jak pole pod krzywą ROC (AUC) i miara F1 o około 5–8 punktów procentowych. Co ważne, te ulepszenia nie odbywały się kosztem przejrzystości: model generuje mapy uwagi z TabNet oraz mapy aktywacji z CNN, które podkreślają, które geny i regiony miały największy wpływ na każdą prognozę.
Równoważenie dokładności, prywatności i zaufania
Ponieważ dane genomowe są głęboko osobiste, autorzy zbadali też sposoby ochrony prywatności przy zachowaniu użytecznego sygnału. Wprowadzili adaptacyjny mechanizm prywatności, który dodaje więcej szumu do cech wysoce wrażliwych, a mniej do pozostałych, w połączeniu z maskowaniem wybranych wejść. Testy wykazały, że nawet przy umiarkowanym poziomie szumu model zachowywał wysoką dokładność i zdolność rozróżniania, a wydajność pogarszała się stopniowo w miarę zaostrzania ochrony. Równocześnie interpretowalne wzorce uwagi i aktywacji często wskazywały na geny już znane z ról w nowotworach i regulacji układu odpornościowego, co sugeruje, że system nie tylko zapamiętuje dane, lecz wydobywa biologicznie istotne sygnały. Badanie ablacjne — systematyczne usuwanie części architektury — potwierdziło, że każdy moduł, szczególnie warstwa AFR, wnosi mierzalny wkład w wydajność.
Co to oznacza dla przyszłej medycyny
Mówiąc prosto, praca ta oferuje inteligentniejszy sposób przesiewania ogromnych arkuszy danych genomowych w poszukiwaniu wzorców powiązanych z chorobami, jednocześnie pokazując, które wpisy w tych arkuszach miały największe znaczenie. Łącząc ukierunkowany wybór cech, staranne rafinowanie i rozpoznawanie wzorców, hybrydowy model poprawia trafność prognoz, pozostaje obliczeniowo wykonalny i dostarcza wizualnych wskazówek, które klinicyści i biolodzy mogą interpretować. Choć potrzebne są dalsze testy na szerszych i bardziej zróżnicowanych grupach pacjentów, takie ramy mogą pomóc w identyfikacji nowych biomarkerów, doprecyzowaniu podtypów chorób i wspierać narzędzia decyzyjne w medycynie precyzyjnej — przybliżając analizę DNA przez AI do rzeczywistego zastosowania.
Cytowanie: Swain, M.K., Kamila, N.K., Jena, L. et al. Hybrid deep learning framework for accurate classification of high dimensional genomic data. Sci Rep 16, 5919 (2026). https://doi.org/10.1038/s41598-026-36128-7
Słowa kluczowe: głębokie uczenie genomowe, odkrywanie biomarkerów nowotworowych, interpretowalna sztuczna inteligencja, medycyna precyzyjna, genomika z zachowaniem prywatności