Clear Sky Science · pl
Ilustrowane ilościowo badanie związków cytotoksycznych z wykorzystaniem deskryptorów grafowych i uczenia maszynowego
Dlaczego to badanie ma znaczenie dla przyszłych leków przeciwnowotworowych
Leki przeciwnowotworowe zabijające komórki nowotworowe, zwane lekami cytotoksycznymi, często balansują między ratowaniem życia a wywoływaniem poważnych skutków ubocznych. Aby projektować bezpieczniejsze i skuteczniejsze terapie, naukowcy potrzebują szybkich i wiarygodnych metod przewidywania, jak te leki przemieszczają się w organizmie — jak dobrze są wchłaniane, jak łatwo przenikają przez błony komórkowe i gdzie ostatecznie się kumulują. To badanie pokazuje, że matematyczne opisy cząsteczek, połączone z nowoczesnym uczeniem maszynowym, mogą dokładnie oszacować kluczową właściwość kontrolującą to zachowanie, co może przyspieszyć poszukiwanie lepszych terapii przeciwnowotworowych.
Kluczowa powierzchnia, która kontroluje, dokąd leki mogą dotrzeć
Jednym z centralnych pojęć w pracy jest topologiczna polarna powierzchnia, czyli Top_PSA. W prostych słowach, jest to liczba odzwierciedlająca, jaka część powierzchni cząsteczki składa się z regionów „polarnych” — fragmentów preferujących wodę i zdolnych do tworzenia wiązań wodorowych. Cząsteczki o bardzo wysokiej polarnej powierzchni często mają trudności z przechodzeniem przez tłuszczowe błony komórkowe i mogą być słabo wchłaniane po podaniu doustnym. Cząsteczki o bardzo niskiej polarnej powierzchni mogą natomiast zbyt łatwo przenikać bariery, czasem powodując niepożądane działania w wrażliwych tkankach, takich jak mózg. Top_PSA stała się popularnym skrótem do szacowania tych właściwości transportowych, ponieważ można ją szybko obliczyć z dwuwymiarowego rysunku cząsteczki, bez potrzeby kosztownych symulacji 3D.
Przekształcanie rysunków molekularnych w liczby
Naukowcy zgromadzili skuratowaną kolekcję 156 różnych związków cytotoksycznych pochodzących z rzeczywistych leków przeciwnowotworowych i związków eksperymentalnych. Następnie przekształcili każdą cząsteczkę w 58 tak zwanych deskryptorów — liczb opisujących cechy takie jak liczba atomów, liczba pierścieni, elastyczność wiązań, liczba atomów zdolnych do tworzenia wiązań wodorowych czy stopień polarności i elektroujemności różnych fragmentów. Wiele z tych deskryptorów pochodzi z teorii grafów, gdzie cząsteczkę traktuje się jako sieć połączonych węzłów i krawędzi. Ten bogaty numeryczny portret każdej cząsteczki posłużył jako wejście do modeli komputerowych próbujących przewidzieć wartości Top_PSA obliczane przez powszechnie używane narzędzia chemiczne.
Testowanie różnych dróg do dokładnego przewidywania
Aby znaleźć najlepszy sposób powiązania deskryptorów z Top_PSA, zespół porównał kilka strategii modelowania. Wypróbowali standardową regresję liniową oraz dwie „uregularyzowane” wersje zwane regresją ridge i LASSO, które lepiej radzą sobie z hałaśliwymi, nakładającymi się informacjami. Zbadali też różne schematy przygotowania danych: dopasowywanie modeli bezpośrednio do surowych deskryptorów, kompresję za pomocą analizy składowych głównych (PCA), skalowanie w sposób zmniejszający wpływ wartości skrajnych (robust scaling), korygowanie wartości odstających oraz przycinanie silnie skorelowanych cech przy użyciu miary zwanej współczynnikiem inflacji wariancji. Każde podejście oceniano starannie przy użyciu walidacji krzyżowej k‑fold, metody, która wielokrotnie dzieli dane na zbiory treningowe i testowe, aby zabezpieczyć się przed przeuczeniem.
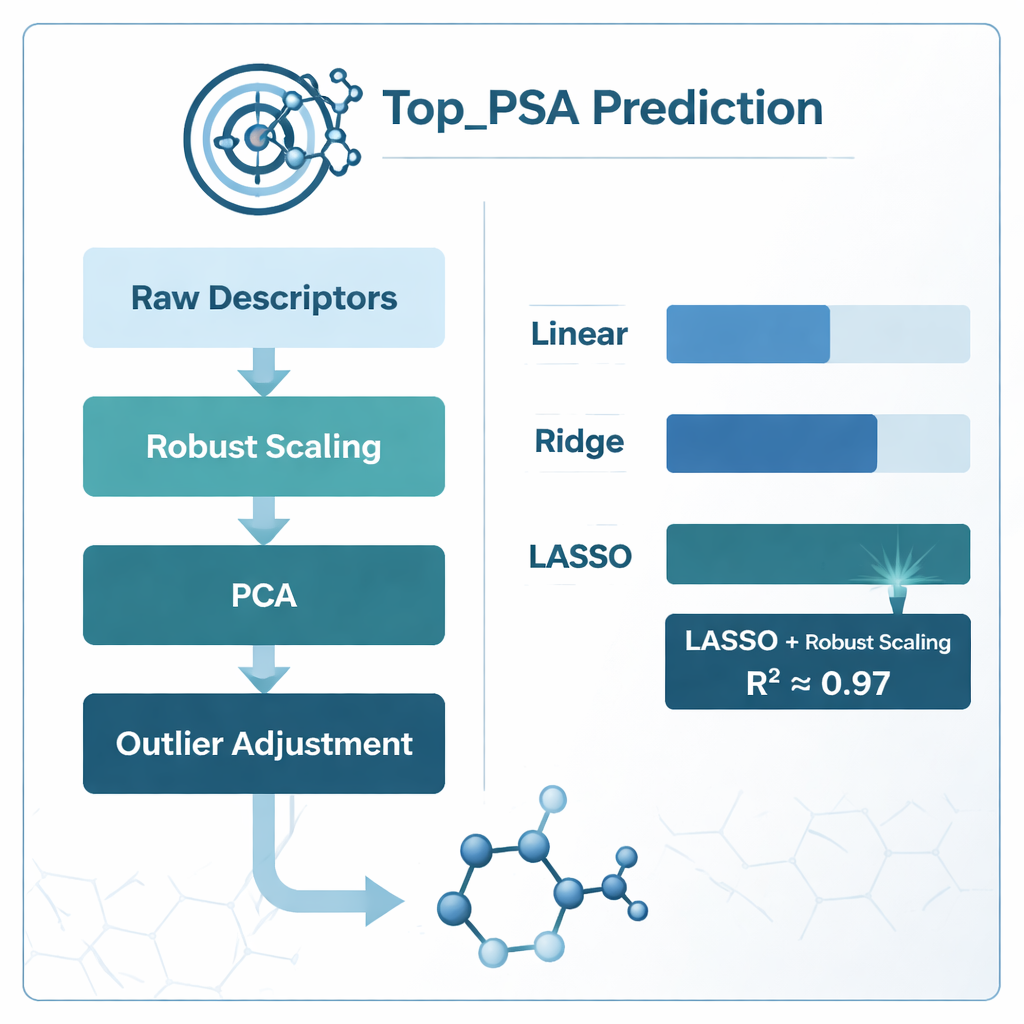
Co zadziałało najlepiej i czego nauczyły się modele
Wyraźnym zwycięzcą okazało się połączenie skalowania odpornego (robust scaling) z regresją LASSO, które osiągnęło współczynnik determinacji (R²) około 0,97 — oznacza to, że model potrafił wyjaśnić około 97% zmienności Top_PSA wśród 156 związków. Modele oparte na PCA zbliżały się pod względem surowej dokładności, ale były trudniejsze do chemicznej interpretacji, ponieważ oryginalne deskryptory były zmieszane w abstrakcyjne składowe. Proste przycinanie skorelowanych deskryptorów za pomocą współczynnika inflacji wariancji w rzeczywistości pogorszyło wydajność, co sugeruje, że niektóre nakładające się miary wciąż niosą użyteczną informację chemiczną. Analizując, które wagi deskryptorów pozostały niezerowe w LASSO, autorzy stwierdzili, że najważniejsze czynniki to obecność heteroatomów, takich jak azot i tlen, zdolność do oddawania i przyjmowania wiązań wodorowych oraz indeksy śledzące rozmieszczenie atomów o różnej elektroujemności w grafie molekularnym — wszystkie cechy zgodne z intuicyjnym chemicznym rozumieniem polarnej powierzchni.
Jak to może ukierunkować lepsze projektowanie leków
Dla czytelników spoza dziedziny kluczowy przekaz jest taki, że starannie przygotowane matematyczne „odciski palców” cząsteczek, w połączeniu z dobrze dobranymi metodami uczenia maszynowego, mogą dawać szybkie i wiarygodne oszacowania tego, jak „lepne” lub „śliskie” będą leki przeciwnowotworowe w trakcie podróży przez organizm. Badanie dostarcza praktycznych wskazówek dla innych badaczy, jak przygotowywać dane deskryptorowe, które podejścia modelowe faworyzować i których skrótów unikać. W dłuższej perspektywie takie odporne, interpretowalne modele Top_PSA mogą pomóc chemikom odfiltrowywać ogromne wirtualne biblioteki potencjalnych leków, koncentrując wysiłki na związkach o właściwej równowadze między przenikaniem przez błony a bezpieczeństwem — istotny krok w kierunku skuteczniejszych i mniej toksycznych terapii przeciwnowotworowych.
Cytowanie: Ahmad, S., Javed, S., Khalid, S. et al. A quantitative study of cytotoxic compounds using graph based descriptors and machine learning. Sci Rep 16, 5076 (2026). https://doi.org/10.1038/s41598-026-35728-7
Słowa kluczowe: leki cytotoksyczne, polarna powierzchnia, deskryptory molekularne, uczenie maszynowe, przepuszczalność leków