Clear Sky Science · pl
Zakłócenie wewnątrzkomórkowej homeostazy żelaza przez dysfunkcję mitochondriów związaną z tłumieniem ekspresji ATP13A2
Dlaczego żelazo wewnątrz komórek mózgu ma znaczenie
Choroba Parkinsona najczęściej kojarzy się z drżeniem i sztywnymi ruchami, ale głęboko wewnątrz zajętych komórek mózgu rozwija się inny dramat: żelazo, niezbędny metal, zaczyna gromadzić się tam, gdzie nie powinno. W niniejszym badaniu postawiono proste, lecz istotne pytanie: jak dochodzi do tego nagromadzenia żelaza i w jaki sposób może ono uszkadzać maleńkie elektrownie i centra recyklingu w komórkach nerwowych? Odpowiadając, praca dostarcza wskazówek, dlaczego określone rejony mózgu ulegają degeneracji w chorobie Parkinsona i pokrewnych schorzeniach, oraz wskazuje nowe kierunki terapii wykraczające poza zastępowanie dopaminy.
Bliższe spojrzenie na rzadką genetyczną wskazówkę
Naukowcy skupili się na rzadkiej dziedzicznej postaci choroby Parkinsona, zwanej PARK9, wywołanej mutacjami w genie ATP13A2. Gen ten koduje białko występujące w lizosomach — komórkowych kompartmentach odpowiedzialnych za przetwarzanie odpadów i recykling. Osoby z mutacjami ATP13A2 mogą także rozwinąć stan charakteryzujący się odkładaniem żelaza w mózgu. To powiązanie uczyniło ATP13A2 idealnym punktem startowym do zbadania, jak zaburza się równowaga żelaza. Wykorzystując ludzką linię komórek o cechach neuronów, nadprodukującą białko alfa‑synukleinę związaną z Parkinsonem, zespół zastosował krótkie fragmenty RNA do obniżenia ekspresji ATP13A2, a następnie śledził zmiany w dystrybucji żelaza, produkcji energii i kondycji komórek.
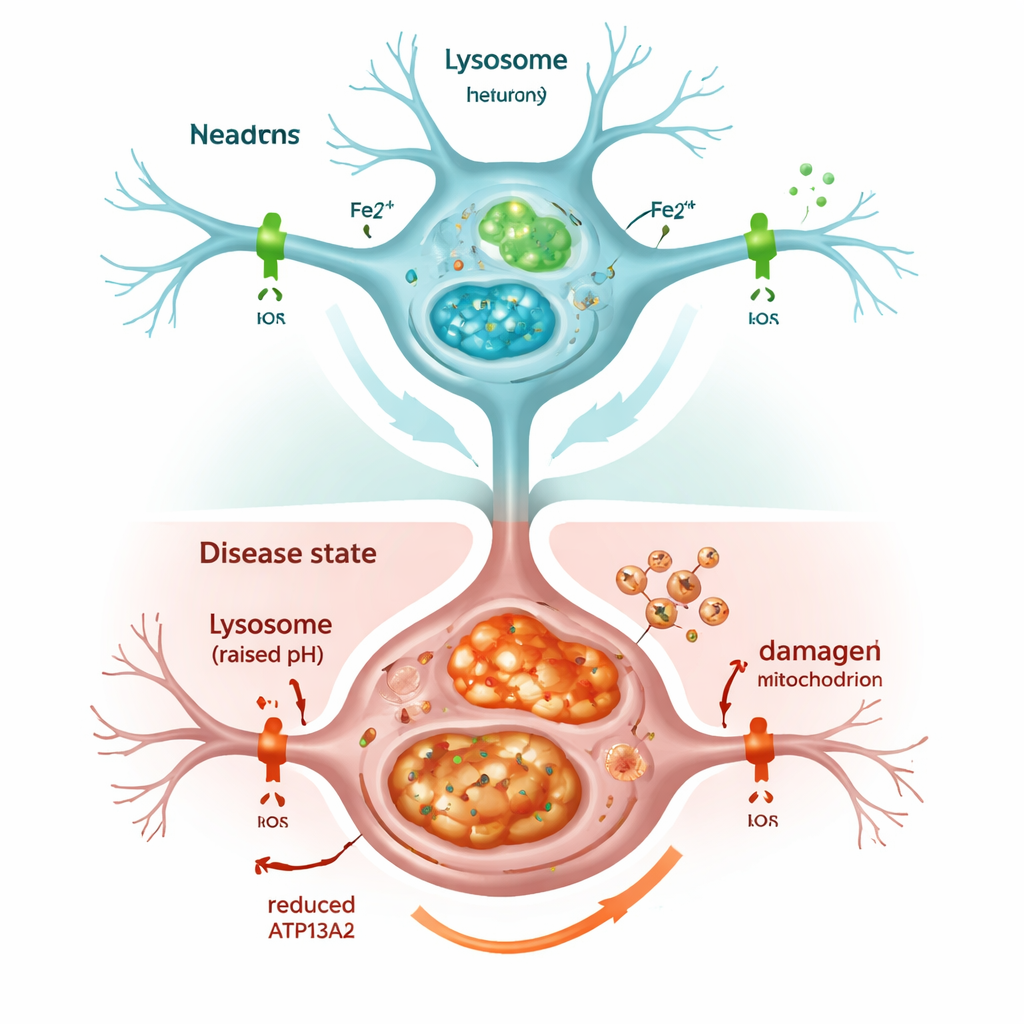
Gdy system recyklingu komórki zatyka się
Wyłączenie ATP13A2 szybko osłabiło funkcję lizosomów. Ich wewnętrzna kwasowość, kluczowa dla rozkładu niechcianych materiałów, spadła, a markery procesu oczyszczania komórkowego, zwanego autofagią, nagromadziły się zamiast być usunięte. W efekcie gromadziła się alfa‑synukleina, co odzwierciedla obserwacje w mózgach osób z Parkinsonem. Komórki wykazały też ogólny wzrost zawartości żelaza, a zwłaszcza wzrost reaktywnie czynnej formy Fe2+ zarówno w lizosomach, jak i w mitochondriach. Komórka reagowała zwiększoną produkcją ferrytyny, białka magazynującego żelazo, ale to nie wystarczało, by zapobiec problemom: przeciążone mitochondria wytwarzały nadmiar reaktywnych form tlenu, a przeżywalność komórek spadała. Leczenie komórek lekiem wiążącym żelazo, podobnym do stosowanych klinicznie, zmniejszyło ten stres oksydacyjny i częściowo przywróciło przeżywalność, podkreślając, że nadmiar żelaza sam w sobie był kluczowym czynnikiem uszkodzeń.
Czujniki żelaza przestają słuchać metalu
Zazwyczaj komórki dysponują systemem sprzężenia zwrotnego, który wykrywa wzrost poziomu żelaza i odpowiada obniżeniem importu żelaza. Białko IRP2 wyczuwa żelazo między innymi poprzez sygnał zależny od hemu pochodzący z mitochondriów, a następnie reguluje produkcję białek przenoszących żelazo na powierzchni komórki. W komórkach pozbawionych ATP13A2 ten mechanizm zawiódł. Transportery wprowadzające żelazo do komórki pozostawały na wysokim poziomie, mimo że żelazo było już podwyższone. Poziomy białka IRP2 zmieniały się niewiele, a dodanie zewnętrznego żelaza nie wywołało jego normalnego rozpadu. Zespół przypisał tę usterkę mitochondriom: uszkodzone mitochondria oddychały mniej wydajnie, wykazywały cechy wadliwego mechanizmu kontroli jakości (mitofagii) i, co kluczowe, straciły zdolność do syntezy hemu — żelazo‑zawierającej cząsteczki, która pomaga IRP2 wyczuwać żelazo. Bez wystarczającej ilości hemu IRP2 nie otrzymywało sygnału „za dużo żelaza” i pozwalało na dalszy napływ żelaza.
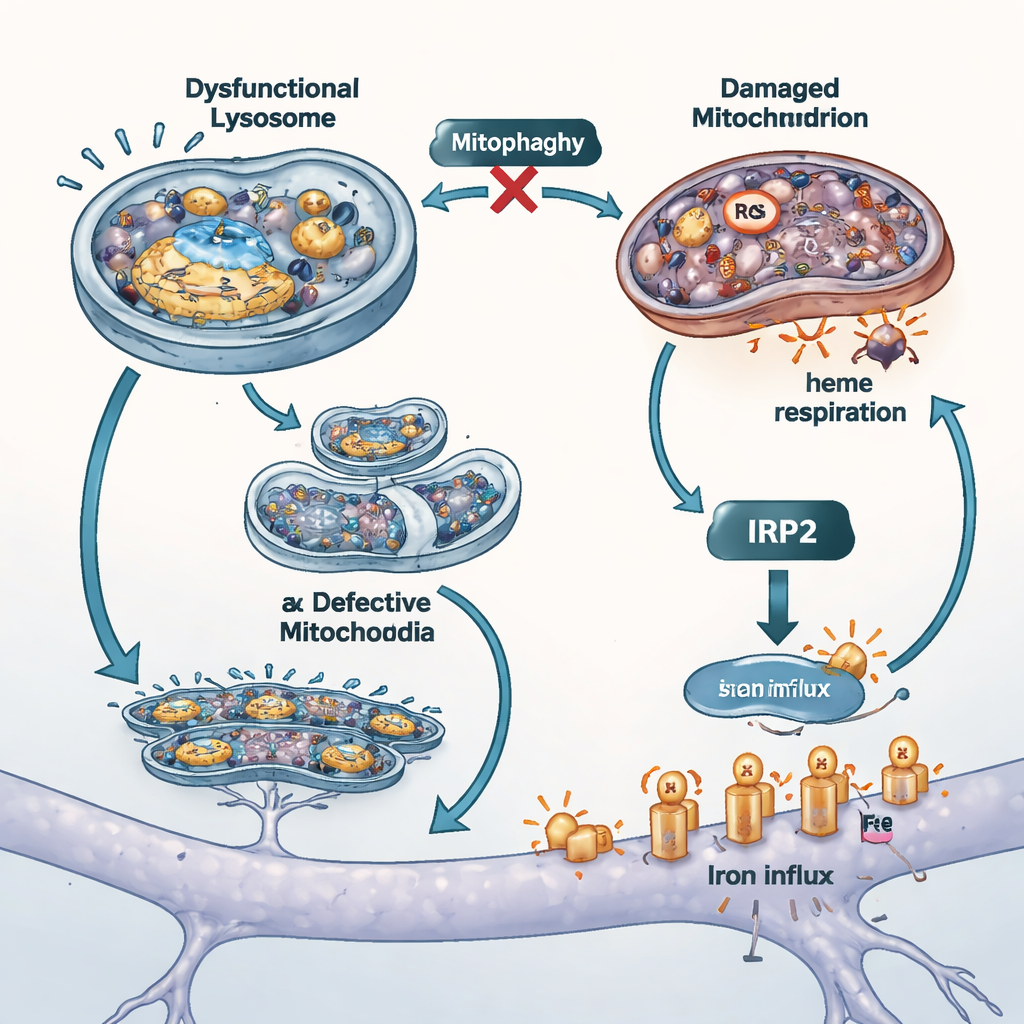
Zamknięcie kranu żelaza i testy w innych modelach
Aby sprawdzić, jak bardzo niekontrolowany napływ żelaza przyczynia się do uszkodzeń komórek, naukowcy zablokowali dwa główne szlaki jego pobierania. Użyli bezżelaznej wersji białka krwi transferyny, aby konkurować o jeden importer, oraz małego leku hamującego aktywność drugiego transportera, DMT1. Obie interwencje obniżyły ogólne i wolne żelazo w komórkach, zmniejszyły stres oksydacyjny w mitochondriach i poprawiły przeżywalność, sugerując, że kanały żelazowe na powierzchni są istotnymi wzmacniaczami uszkodzeń przy utracie ATP13A2. Badacze powtórzyli też kluczowe eksperymenty w komórkach pozbawionych innego genu związanego z Parkinsonem, PINK1, znanego z zaburzenia mitofagii. Te komórki wykazały podobne połączenie nagromadzenia żelaza i osłabionej produkcji hemu, co wspiera koncepcję, że kontrola jakości mitochondriów i równowaga żelaza są ściśle powiązane w różnych formach choroby.
Co to oznacza dla choroby Parkinsona i przyszłych terapii
Mówiąc wprost, badanie przedstawia błędne koło. Gdy ATP13A2 jest tłumione, lizosomy nie radzą sobie z usuwaniem uszkodzonych składników, w tym wadliwych mitochondriów. Osłabione mitochondria wytwarzają mniej energii i mniej hemu, co tłumi komórkowy system wykrywania żelaza. Żelazo nadal napływa przez transportery powierzchniowe, gromadzi się w wrażliwych kompartmentach i napędza toksyczne reakcje, które dodatkowo uszkadzają mitochondria. Z czasem ta pętla może pomóc wyjaśnić, dlaczego niektóre neurony obumierają w chorobie Parkinsona i pokrewnych zaburzeniach związanych z odkładaniem żelaza w mózgu. Wyniki sugerują, że przyszłe terapie mogłyby nie tylko usuwać nadmiar żelaza, lecz także przywracać prawidłową funkcję lizosomów, kontrolę jakości mitochondriów i produkcję hemu — atakując problem u źródła, zamiast jedynie usuwać nadmiar metalu po fakcie.
Cytowanie: Murakami, T., Ohuchi, K., Kiuchi, M. et al. Disruption of intracellular iron homeostasis through mitochondrial dysfunction associated with suppression of ATP 13A2 expression. Sci Rep 16, 5007 (2026). https://doi.org/10.1038/s41598-026-35368-x
Słowa kluczowe: Choroba Parkinsona, żelazo w mózgu, mitochondria, lizosomy, synteza hemu