Clear Sky Science · pl
Badanie funkcjonalnych reszt „hot-spot” enzymu przez monitorowanie reakcji enzymatycznej w czasie rzeczywistym za pomocą NMR i metod obliczeniowych
Dlaczego to ma znaczenie dla przyszłych leków przeciwwirusowych
Favipirawir to tabletka stosowana już przeciw grypie i badana w kontekście COVID-19, ale nie działa w postaci, którą połykamy. Nasze własne komórki muszą najpierw przekształcić go w aktywną cząsteczkę blokującą wirusy. W tym badaniu rozłożono niemal atom po atomie, jak jeden ludzki enzym przeprowadza kluczowy etap aktywacji i które drobne fragmenty enzymu pełnią rolę „hot-spotów” kontrolujących szybkość i skuteczność włączenia leku. Zrozumienie tych szczegółów może ukierunkować projektowanie następnej generacji leków przeciwwirusowych, które będą zarówno silniejsze, jak i bardziej przewidywalne u pacjentów.
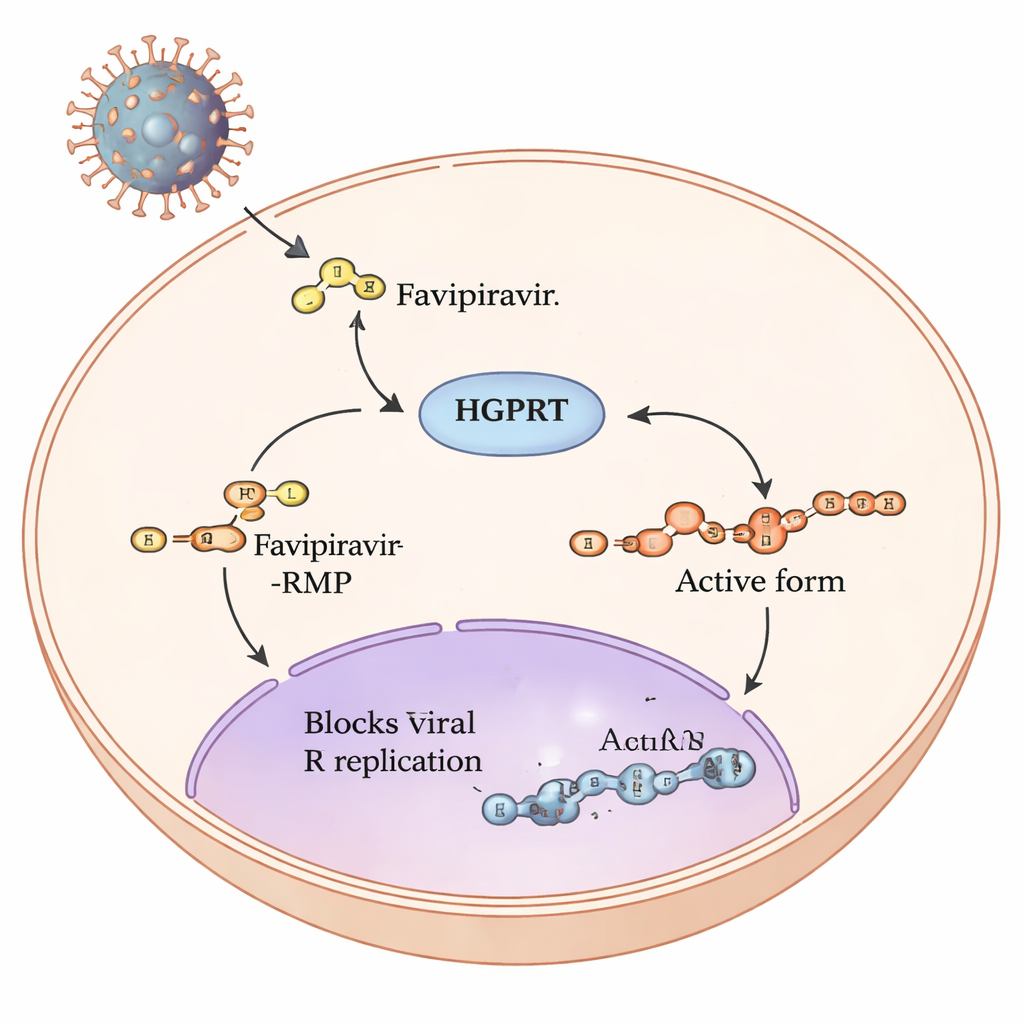
Podróż proleku wewnątrz naszych komórek
Favipirawir to tzw. prolek: po wejściu do komórek ludzkich szereg reakcji chemicznych przebudowuje go do postaci, która może zablokować mechanizmy kopiowania wirusowego RNA, takie jak u wirusa grypy czy SARS‑CoV‑2. Pierwszy i najwolniejszy etap tej drogi przeprowadza ludzki enzym zwany hipoksantyna‑guanina fosforybozylotransferazą, czyli HGPRT. HGPRT dołącza do favipirawiru małą grupę cukrowo‑fosforanową, tworząc favipirawir‑RMP. Dopiero po tym kroku inne enzymy mogą zbudować w pełni aktywną formę trifosforanową, która bezpośrednio zakłóca polimerazę RNA wirusa. Ponieważ pierwszy etap zależny od HGPRT działa jak wąskie gardło decydujące o ilości powstającego aktywnego leku, autorzy postanowili określić, które fragmenty HGPRT są kluczowe dla przetwarzania favipirawiru.
Obserwowanie chemii w czasie rzeczywistym za pomocą NMR
Wyjątkowo favipirawir zawiera atom fluoru, który zachowuje się jak mały nadajnik radiowy w polu magnetycznym. Zespół wykorzystał to, stosując spektroskopię magnetycznego rezonansu jądrowego fluoru‑19, aby na żywo śledzić, ile favipirawiru i ile favipirawir‑RMP znajduje się w probówce w miarę przebiegu reakcji. Ponieważ jedynie lek zawiera fluor, sygnały NMR są czyste i łatwe do śledzenia. Poprzez wielokrotne rejestrowanie widm przez 12 godzin badacze mogli obserwować zanikanie leku wyjściowego i narastanie produktu zmodyfikowanego, a następnie wyciągać standardowe miary kinetyczne, takie jak szybkość reakcji i pozorna siła wiązania enzymu z lekiem.
Dostrajanie kluczowych pozycji w enzymie
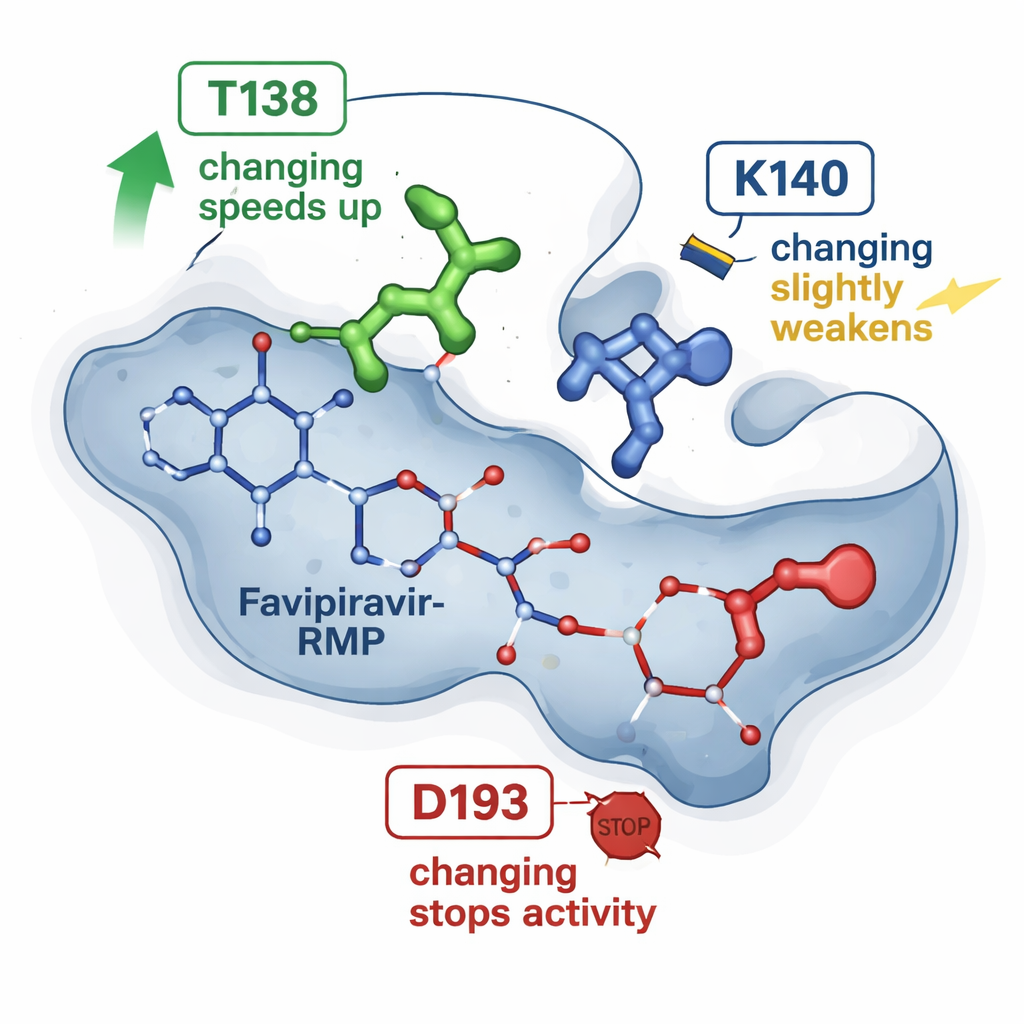
Wcześniejsze migawki rentgenowskie HGPRT związanej z favipirawir‑RMP sugerowały kilka aminokwasów, które otaczają lek w kieszeni białkowej. Nowa praca testuje trzy z tych pozycji, wprowadzając precyzyjne zmiany jednego aminokwasu i porównując każdy mutant z naturalnym enzymem. Zmiana nazwana T138A niespodziewanie zwiększyła ponad czterokrotnie do sześciokrotnie szybkość przekształcania favipirawiru, mimo że usunęła grupę chemiczną, która uważana była za pomocną w utrzymywaniu leku. Druga zmiana, K140M, nieznacznie spowolniła reakcję i nieco osłabiła pozorne wiązanie. Trzecia zmiana, D193N, całkowicie zlikwidowała zdolność enzymu do tworzenia favipirawir‑RMP, choć zmienione białko nadal mogło być produkowane i wiązać produkt. Razem te wyniki pokazują, że nie wszystkie punkty styku są równe: niektóre pełnią rolę subtelnych regulatorów szybkości, podczas gdy inne są niezbędnymi przełącznikami.
Symulowanie ruchomych części na komputerze
Aby wyjść poza statyczne struktury, badacze sięgnęli po symulacje molekularne. Wyjściowo korzystając ze znanej trójwymiarowej struktury HGPRT z favipirawir‑RMP, użyli ustalonych narzędzi obliczeniowych do oszacowania, jak silnie lek wiąże się w każdym mutancie oraz do uruchomienia wielu krótkich symulacji dynamiki molekularnej. Symulacje te śledzą, jak atomy drgają i oddziałują przez dziesiątki nanosekund. Obliczenia zgadzały się z obserwacjami z NMR: wariant T138A miał tendencję do korzystniejszego utrzymywania favipirawir‑RMP, ale też pokazywał epizody, w których lek przemieszczał się w kierunku „ścieżki ucieczki”, kierowany przez inną resztę (K140), która chwilowo kotwiczy grupę fosforanową przed uwolnieniem. W przeciwieństwie do tego wariant D193N nadal trzymał produkt, lecz najprawdopodobniej zawodził na wcześniejszym etapie katalitycznym wymagającym jonów magnezu, co wyjaśnia utratę aktywności mimo stabilnego wiązania.
Mapa drogowa do mądrzejszego projektowania leków przeciwwirusowych
Łącząc pomiary NMR w czasie rzeczywistym z szczegółowymi modelami komputerowymi, to badanie mapuje funkcjonalne hot-spoty w HGPRT, które decydują o tym, jak efektywnie favipirawir jest aktywowany. Dla osób niebędących specjalistami wniosek jest taki, że nasze własne enzymy mogą w dużym stopniu wpływać na ilość aktywnego leku przeciwwirusowego gromadzonego w komórkach, i że zmiana kształtu leku lub kieszeni enzymu może dramatycznie zmienić ten rezultat. Hybrydowa strategia autorów stanowi ogólny plan badania, jak inne leki wchodzą w interakcje ze swoimi białkowymi celami, co może przyspieszyć rozwój nowych związków przeciwwirusowych lepiej dopasowanych do mechanizmów aktywacyjnych w organizmie.
Cytowanie: Sugiki, T., Yoshida, T., Tsukamoto, M. et al. Investigation of the functional hot-spot residues of an enzyme by real-time monitoring of the enzymatic reaction using NMR and computational approaches. Sci Rep 16, 5896 (2026). https://doi.org/10.1038/s41598-026-35354-3
Słowa kluczowe: favipirawir, aktywacja przeciwwirusowa, enzym HGPRT, spektroskopia NMR, projektowanie leków