Clear Sky Science · pl
Ocena biochemicznych podstaw odporności na ogólnoustrojową amyloidozę
Kiedy drobne zmiany w białku blokują śmiertelne nagromadzenie
Wiele przewlekłych chorób zapalnych, od reumatoidalnego zapalenia stawów po gruźlicę, może wywołać rzadkie, lecz często śmiertelne powikłanie zwane ogólnoustrojową amyloidozą AA. W tym stanie normalne białko krwi odkłada się w postaci sztywnych włókien, które zatykają narządy. Badanie stawia zaskakująco optymistyczne pytanie: czy niewielkie, naturalne zmiany w tym białku mogą sprawić, że niektóre zwierzęta są w dużej mierze odporne na chorobę — i jeśli tak, to jak?
Ukryte zagrożenie nagromadzeń białkowych
Amyloidoza AA rozpoczyna się od zapalnego sygnału alarmowego we krwi zwanego surowiczym amyloidem A (SAA). Podczas silnego lub długotrwałego zapalenia poziomy SAA mogą wzrosnąć tysiąckrotnie w stosunku do normy. U niektórych ludzi i zwierząt fragment tego białka fałduje się nieprawidłowo i układa w długie włókna, zwane włóknami amyloidowymi, które rozprzestrzeniają się po narządach, takich jak śledziona i nerki. Z czasem włókna te osłabiają funkcję narządów. Jednak nie każdy osobnik z wysokim poziomem SAA rozwija amyloidozę, a niektóre szczepy myszy pozostają zaskakująco odporne nawet przy laboratoryjnie wywołanym nasileniu choroby. Zrozumienie przyczyn tej odporności może wskazać nowe strategie zapobiegania odkładaniu amyloidu u ludzi.
Odporne myszy i ich szczególne wersje białka
U myszy większość włókien amyloidowych AA powstaje z jednej wersji SAA nazwanej SAA1.1, która jest silnie związana z chorobą. Jednak niektóre szczepy myszy głównie produkują nieco zmienione warianty, nazwane SAA1.5 i SAA2.2, i te szczepy rzadko rozwijają ogólnoustrojową amyloidozę AA. Białka różnią się zaledwie kilkoma cegiełkami budulcowymi (aminokwasami), lecz te różnice skupiają się w ciasno upakowanym regionie tworzącym wewnętrzne jądro patologicznych włókien. Badacze wysunęli hipotezę, że te drobne różnice nie uniemożliwiają ogólnego agregowania się białek, lecz zapobiegają przyjęciu przez nie tej konkretnej, szkodliwej formy włókna.
Testy białek w laboratorium
Aby sprawdzić ten pomysł, zespół wyprodukował wszystkie trzy warianty mysiego SAA w bakteriach i obserwował ich zachowanie w doświadczeniach in vitro. Monitorowano tworzenie włókien za pomocą barwnika fluorescencyjnego, który świeci, gdy powstaje amyloid, oraz weryfikowano struktury mikroskopią elektronową. Wersja powiązana z chorobą, SAA1.1, chętnie tworzyła długie, proste włókna. SAA2.2 także mogła tworzyć włókna, lecz były one grubsze, bardziej skręcone i strukturalnie bardziej zróżnicowane, a ich sygnał barwnika był słabszy. SAA1.5, w przeciwieństwie do nich, nie tworzyła włókien w badanych warunkach. Gdy naukowcy dodali maleńkie próbki prawdziwych włókien chorobowych pobranych z chorych śledzion mysich jako „nasiona”, SAA1.1 szybko rosła na nich, kopiuje strukturę oryginału — podobnie jak prion. Co znamienne, SAA1.5 i SAA2.2 w ogóle nie przyłączały się do tych nasion; włókna ex vivo nie były w stanie wciągnąć ich w patogenną formę.
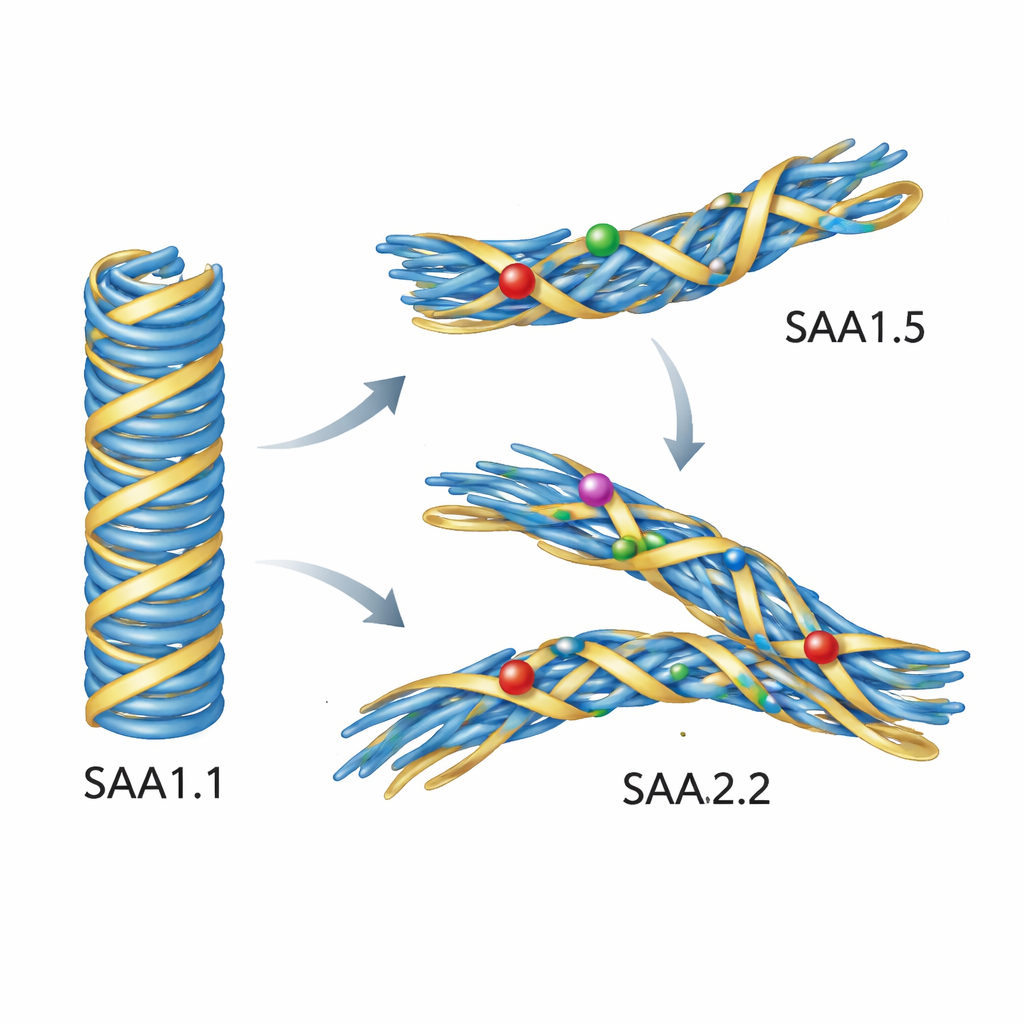
Symulacje ujawniają, dlaczego warianty odrzucają złą formę
Sama praca eksperymentalna nie pokazała dokładnie, co dzieje się na poziomie atomowym, więc autorzy sięgnęli po szczegółowe symulacje komputerowe. Wyszli od wysokorozdzielczej struktury patologicznego włókna AA zbudowanego z SAA1.1, po czym komputerowo podstawili sekwencje SAA1.5 i SAA2.2. Gdy symulowali te włókna w wodzie w temperaturze ciała, model oparty na SAA1.1 pozostał zadziwiająco stabilny. Natomiast włókna z SAA1.5 lub SAA2.2 przesuwały się i deformowały. Kluczowy region pętli w jądrze wysunął się na zewnątrz i poluzował kontakt z początkowym segmentem białka, a kilka bocznych łańcuchów obróciło się do nowych orientacji. Te subtelne przemieszczenia zaburzyły ciasne upakowanie definiujące chorobowy fałd. Innymi słowy, wariantowe sekwencje nie miały problemu z tworzeniem włókien ogólnie — ale nie mieściły się wygodnie w wzorcu patologicznego włókna AA.
Jak rozwiązania natury wskazują kierunki przyszłych terapii
Łącząc wyniki, praca pokazuje, że „odporne na amyloid” szczepy myszy nie są chronione dlatego, że ich SAA w ogóle nie może agregować. Raczej ich wersje SAA są strukturalnie niekompatybilne z jedną, konkretną formą włókna powodującą ogólnoustrojową amyloidozę AA. Białka nadal mogą się zlepiać, ale robią to w alternatywne, pozornie łagodne formy. Podobne ochronne mutacje są znane w innych chorobach związanych z niewłaściwym fałdowaniem białek, w tym w niektórych przypadkach chorób prionowych i choroby Alzheimera. To sugeruje szerszą zasadę: zmodyfikowanie białka skłonnego do choroby tak, by nie mogło przyjąć swojej toksycznej architektury — przy jednoczesnym zachowaniu normalnej funkcji — może wystarczyć, by zapobiec chorobie. W dłuższej perspektywie terapie inspirowane tymi naturalnymi „odpornymi” wariantami, lub krótkimi fragmentami pochodzącymi z nich, mogłyby pomóc kierować białka z dala od szkodliwych form i ku bezpiecznym.
Cytowanie: Moderer, T., Schnell, A.F., Scheurmann, N.J. et al. Assessment of the biochemical basis underlying the resistance against systemic amyloidosis. Sci Rep 16, 1313 (2026). https://doi.org/10.1038/s41598-026-35297-9
Słowa kluczowe: amyloidoza AA, surowiczy amyloid A, niewłaściwe fałdowanie białek, odporność na amyloid, modele mysie