Clear Sky Science · pl
Losowy LASSO dla ekstremalnie wysokowymiarowych danych genomowych
Szukanie igieł w genomowych stogach siana
Współczesna biologia potrafi zmierzyć dziesiątki tysięcy genów jednocześnie, podczas gdy badania kliniczne często obejmują zaledwie kilkaset osób. W tej dysproporcji kryją się niewielkie zbiory genów, które naprawdę mają znaczenie przy przewidywaniu ryzyka choroby lub przeżycia. Artykuł przedstawia „Stochastic LASSO”, metodę statystyczną zaprojektowaną tak, by niezawodnie wydobywać te kluczowe geny z oceanów zaszumionych danych genomowych, nawet gdy genów jest znacznie więcej niż pacjentów.
Dlaczego wybór właściwych genów jest tak trudny
Badacze często polegają na narzędziach takich jak LASSO, które ściągają współczynniki mało istotnych genów w stronę zera, zachowując najbardziej informacyjne. Klasyczne wersje LASSO mają jednak problemy, gdy liczba genów przytłacza liczbę próbek — co jest powszechne w genomice nowotworów. Standardowe LASSO może wybrać co najwyżej tylu genów, ile jest pacjentów, i ma skłonność do pomijania genów o podobnym zachowaniu. Wcześniejsze udoskonalenia dodające dodatkowe kary potrafią poradzić sobie z pewną korelacją, ale mogą też zatarć biologiczne znaczenie, zmuszając powiązane geny do zachowywania się tak, jakby wszystkie wpływały na wynik w tym samym kierunku.
Budowanie czystszych losowych próbek
Obiecującym obejściem jest wielokrotne dopasowywanie LASSO na wielu mniejszych, losowo dobranych podzbiorach genów i łączenie wyników. Jednak takie podejścia „bootstrapowe” wciąż borykają się z trzema problemami: skorelowane geny mogą się znosić wzajemnie, wiele genów jest rzadko lub nigdy próbkowanych, a czysty przypadek sprawia, że ostateczny wybór jest niestabilny. Stochastic LASSO rozwiązuje te problemy bezpośrednio poprzez nowy schemat próbkowania zwany bootstrapowaniem opartym na korelacji. Zamiast wybierać geny zupełnie losowo, faworyzuje te, które są mniej skorelowane z już wybranymi, co daje mniejsze zestawy genów znacznie bardziej niezależnych. Zapewnia też, że każdy gen jest używany tyle samo razy w kolejnych uruchomieniach bootstrapu, dzięki czemu żaden gen nie jest niesprawiedliwie pomijany. 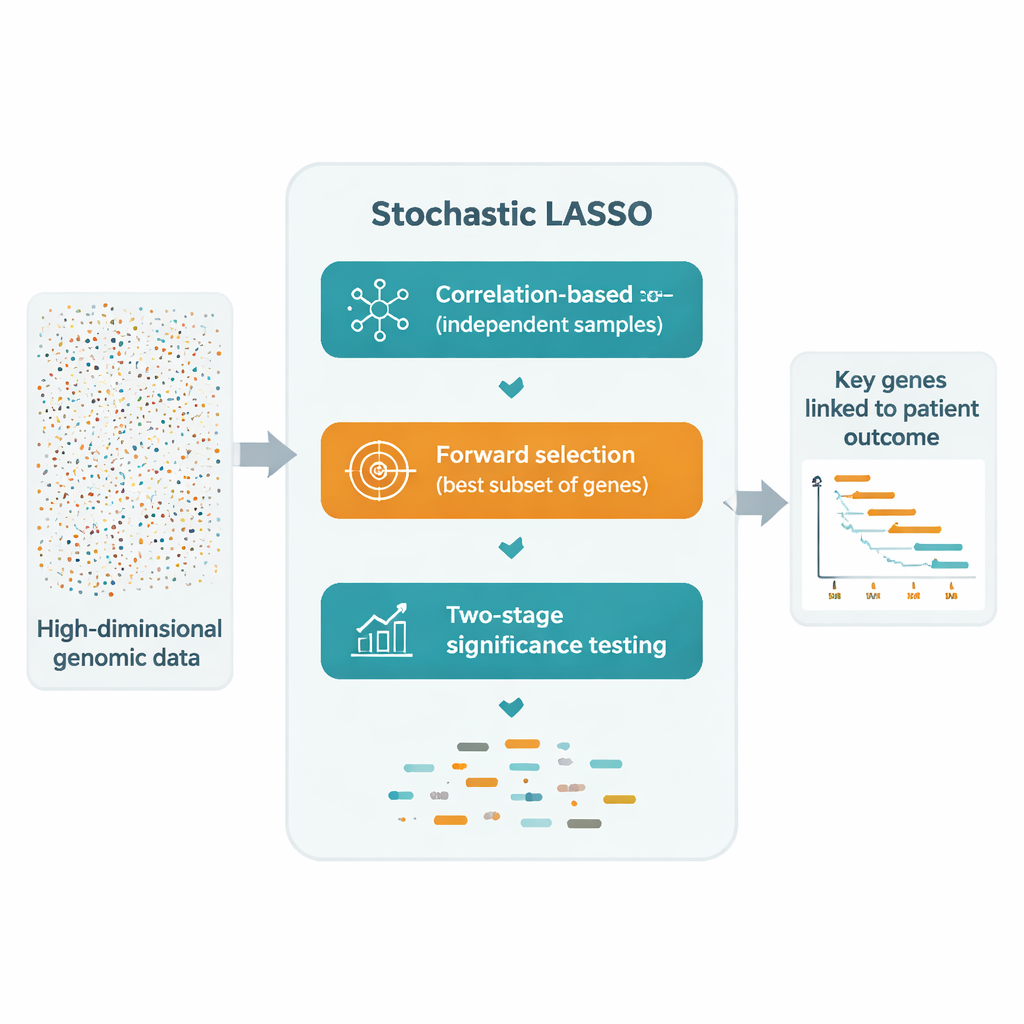
Od lokalnych wskazówek do globalnego zestawu genów
Po zbudowaniu tych czystszych podzbiorów Stochastic LASSO zapisuje, jak duży jest współczynnik każdego genu we wszystkich dopasowaniach bootstrapowych. Ta średnia wartość bezwzględna staje się „lokalnym wynikiem”, który odzwierciedla, jak konsekwentnie dany gen jest ważny. Zamiast testować wyczerpująco wszystkie możliwe kombinacje, metoda konstruuje modele kandydujące, dodając geny w kolejności ich lokalnych wyników i ocenia, jak dobrze każdy kandydat przewiduje wyniki na odrębnych danych walidacyjnych. W ten sposób wybiera kompaktowy zbiór genów, których skumulowane sygnały najlepiej wyjaśniają dane, przy użyciu znacznie mniejszej liczby prób niż tradycyjne metody krokowe.
Testowanie, które geny rzeczywiście mają znaczenie
Aby przejść od „często wybieranych” do „statystycznie przekonujących”, autorzy wprowadzają dwustopniowy test t. Najpierw sprawdzają, czy średni współczynnik każdego genu w bootstrapach jest wyraźnie różny od zera, oznaczając go jako potencjalnie istotny. Następnie, wśród tych kandydatów, pytają, czy efekt konkretnego genu jest większy niż typowy rozmiar efektu wszystkich kandydatów. Tylko geny, które przejdą oba testy, są uznawane za istotne. Ponieważ testy te opierają się na licznych estymatach z bootstrapu, Stochastic LASSO może z pewnością zidentyfikować więcej istotnych genów niż liczba pacjentów — czego konwencjonalne LASSO nie potrafi. 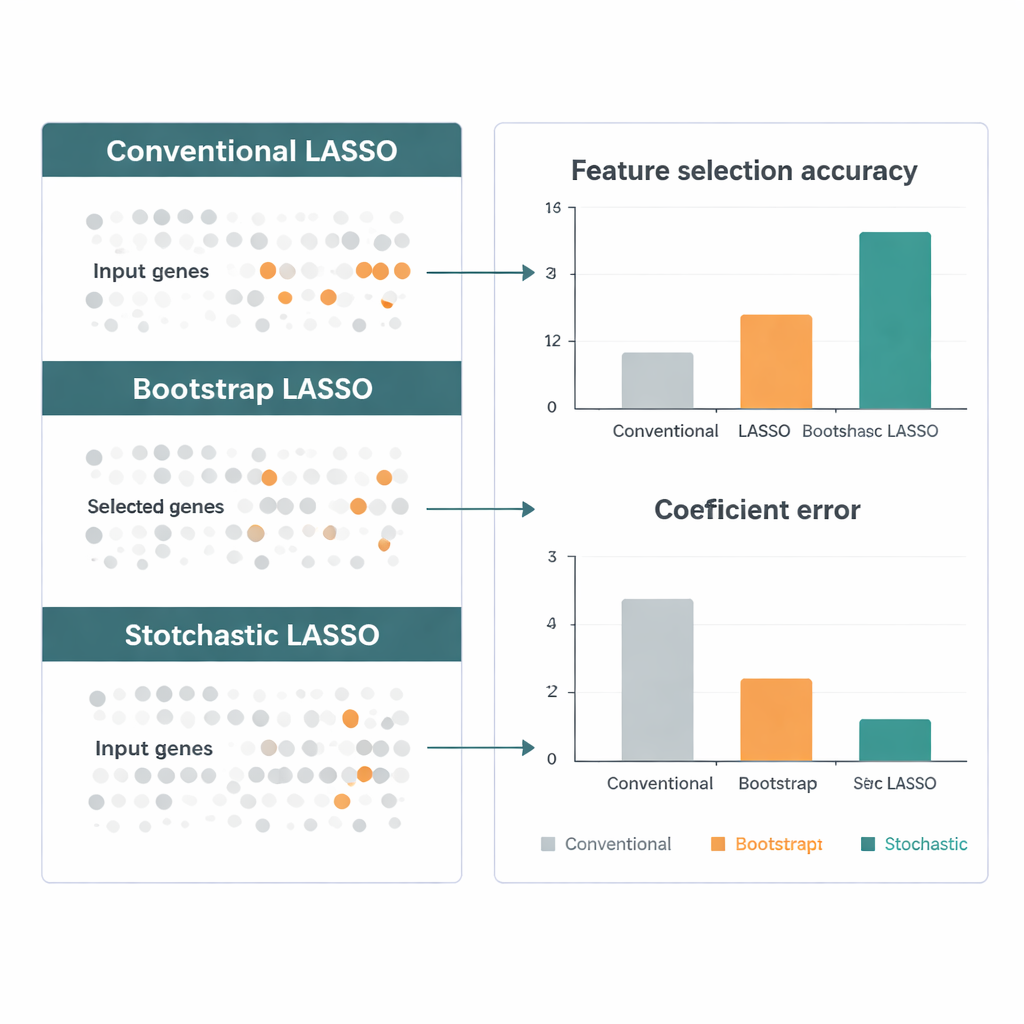
Udowadnianie wartości w symulacjach i danych nowotworowych
Autorzy porównują Stochastic LASSO z kilkoma wiodącymi wariantami LASSO przy użyciu danych symulowanych tak, by naśladować rzeczywiste badania genomowe: bardzo wiele genów, silne korelacje i znane „prawdziwe” sygnały. W różnych scenariuszach nowa metoda częściej odnajduje poprawne geny, dokładniej szacuje ich efekty i pozostaje stabilna między uruchomieniami. Następnie analizują dane z ekspresji genów z The Cancer Genome Atlas dla guzów mózgu, w tym agresywnego glejaka wielopostaciowego. Stochastic LASSO uwydatnia setki genów, których aktywność wiąże się z przeżyciem pacjentów, i wskazuje ścieżki biologiczne — takie jak szlaki sygnalizacyjne i metabolizmu leków — które mają niezależne wsparcie w literaturze, co sugeruje, że metoda jest nie tylko statystycznie precyzyjna, lecz także biologicznie sensowna.
Co to oznacza dla pacjentów i badaczy
Dla osób niebędących specjalistami kluczowy przekaz jest taki, że Stochastic LASSO to inteligentniejszy filtr dla genomowych big data. Pomaga naukowcom oddzielić genu związane z chorobą od statystycznego szumu, nawet gdy dane są ograniczone, a geny silnie powiązane. Dostarczając dokładniejsze i bardziej stabilne listy genów oraz estymaty efektów, może zaostrzyć poszukiwania biomarkerów, celów terapeutycznych i sygnatur prognostycznych w nowotworach i innych złożonych chorobach. Choć zaprezentowano ją przy regresji liniowej, ten sam schemat można wpleść w modele przeżycia i problemy klasyfikacyjne, rozszerzając jej potencjalny wpływ w badaniach biomedycznych.
Cytowanie: Baek, B., Jo, J., Kang, M. et al. Stochastic LASSO for extremely high-dimensional genomic data. Sci Rep 16, 5250 (2026). https://doi.org/10.1038/s41598-026-35273-3
Słowa kluczowe: selekcja cech genomowych, dane wysokowymiarowe, metody LASSO, ekspresja genów w nowotworach, odkrywanie biomarkerów