Clear Sky Science · pl
Kwas mlekowy reguluje oś YTHDF2–FTH1, promując ferroptoza kardiomiocytów i nasilając uszkodzenie przy niedokrwieniu–reperfuzji mięśnia sercowego
Dlaczego pacjenci kardiologiczni powinni się tym przejmować
Kiedy lekarze udrażniają zatkane naczynie wieńcowe po zawale, napływ świeżej krwi ratuje mięsień, ale może też powodować dodatkowe uszkodzenia, znane jako uraz niedokrwienie–reperfuzja. W tym badaniu wykryto zaskakującego winowajcę wewnątrz komórek serca: powszechny produkt metabolizmu — kwas mlekowy. Autorzy pokazują, że kwas mlekowy może przestawić molekularny przełącznik, który skłania komórki serca ku specyficznemu rodzajowi żelazozależnej śmierci komórkowej, pogarszając uszkodzenie. Zrozumienie tej ukrytej ścieżki może wskazać nowe leki lepiej chroniące serce podczas leczenia ratunkowego.
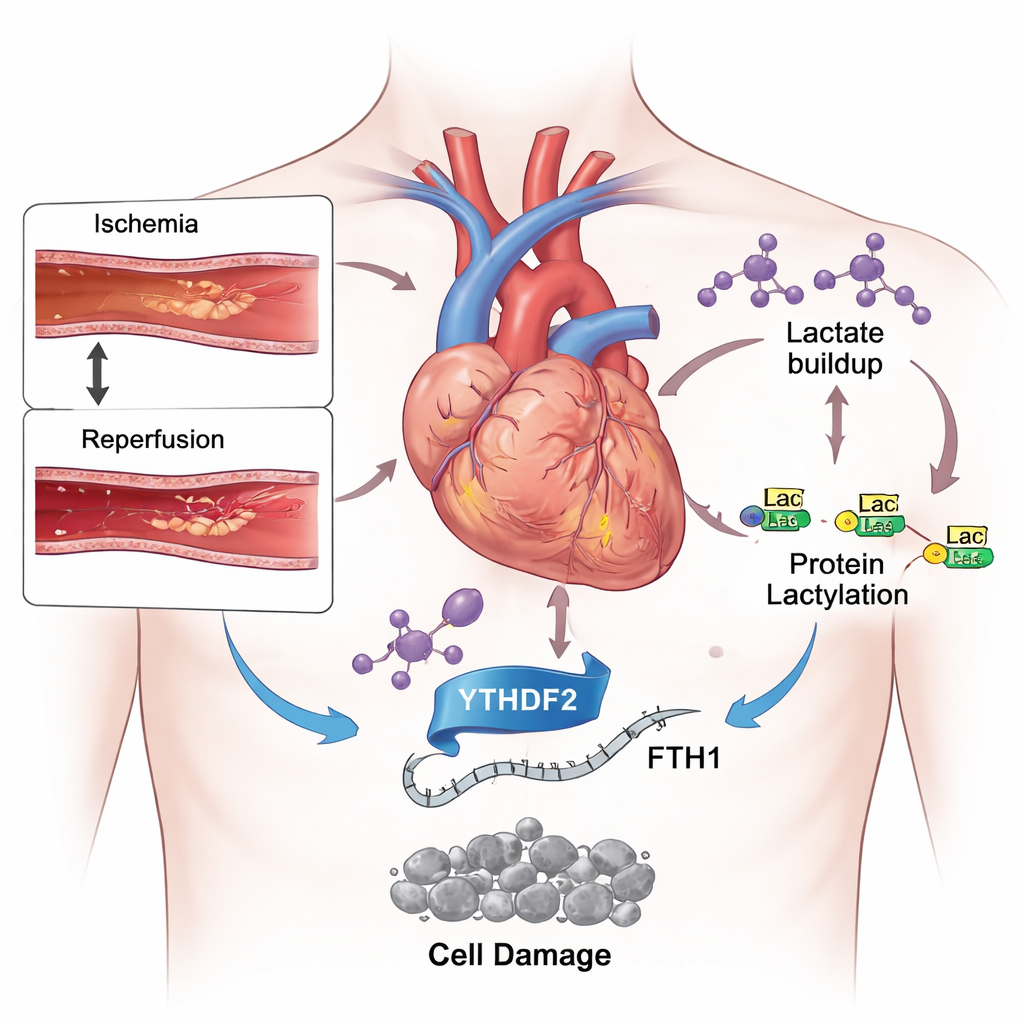
Obosieczny miecz w leczeniu zawału
Współczesna medycyna stała się bardzo skuteczna w szybkim udrażnianiu zatkanych tętnic wieńcowych, ograniczając początkowe uszkodzenie po zawale. Mimo to pacjenci mogą dalej tracić rozległe obszary mięśnia sercowego po przywróceniu przepływu krwi. Jednym z powodów jest to, że nagły powrót tlenu i składników odżywczych wywołuje burzę stresu chemicznego w komórkach serca. Spośród kilku typów śmierci komórkowej uruchamianych w tym kontekście, na uwagę zasługuje nowo poznana — ferroptoza. W przeciwieństwie do bardziej znanych form, takich jak apoptoza, ferroptoza zależy od żelaza i niekontrolowanej oksydacji lipidów w błonach komórkowych, co może trwale osłabić serce.
Jak kwas mlekowy staje się czymś więcej niż „palenie mięśni”
Podczas zawału, głodujący mięsień sercowy przesuwa wykorzystanie paliwa w kierunku glikolizy, systemu awaryjnego, który szybko rozkłada cukier, ale produkuje duże ilości kwasu mlekowego. Używając myszy poddanych krótkotrwałemu zatkaniu i ponownemu otwarciu tętnicy wieńcowej oraz hodowanych komórek przypominających kardiomiocyty wystawionych na niski poziom tlenu, a następnie reoksygenację, badacze stwierdzili gwałtowny wzrost poziomu kwasu mlekowego. Równocześnie wykryli więcej chemicznej modyfikacji zwanej laktylacją na wielu białkach i na histonach — rusztowaniach organizujących DNA. Gdy podawano zwierzętom lek spowalniający glikolizę i zmniejszający produkcję kwasu mlekowego, uszkodzenia serca zmalały, spadły krążące markery urazu, a równowaga między szkodliwym żelazem a ochronnymi antyoksydantami poprawiła się. Wyniki te sugerują, że nadmiar kwasu mlekowego nie jest tylko produktem ubocznym stresu, lecz aktywnym czynnikiem napędzającym uszkodzenia.
Molekularny przełącznik, który poluzowuje kaganiec żelaza
Zagłębiając się dalej, zespół skupił się na YTHDF2 — białku odczytującym chemiczne oznaczenia na RNA i decydującym, jak szybko pewne komunikaty są niszczone. Odkryli, że niedokrwienie–reperfuzja i dodany kwas mlekowy zwiększały poziomy YTHDF2 oraz nasilały laktylację wokół genu, który go koduje, wzmacniając jego produkcję. Jednym z kluczowych celów YTHDF2 okazało się RNA łańcucha ciężkiego ferrytyny (FTH1), istotnej części komórkowej klatki do magazynowania żelaza. FTH1 zwykle chowa żelazo w bezpiecznej formie, zapobiegając jego udziałowi w reaktywnych, uszkadzających reakcjach. W zestresowanych kardiomiocytach YTHDF2 silniej wiązał się z RNA FTH1 i przyspieszał jego rozpad, pozostawiając komórki z mniejszą liczbą ferrytynowych „klatek”, większą ilością żelaza wolnego, nasilonym stresem oksydacyjnym i klasycznymi oznakami ferroptozy.
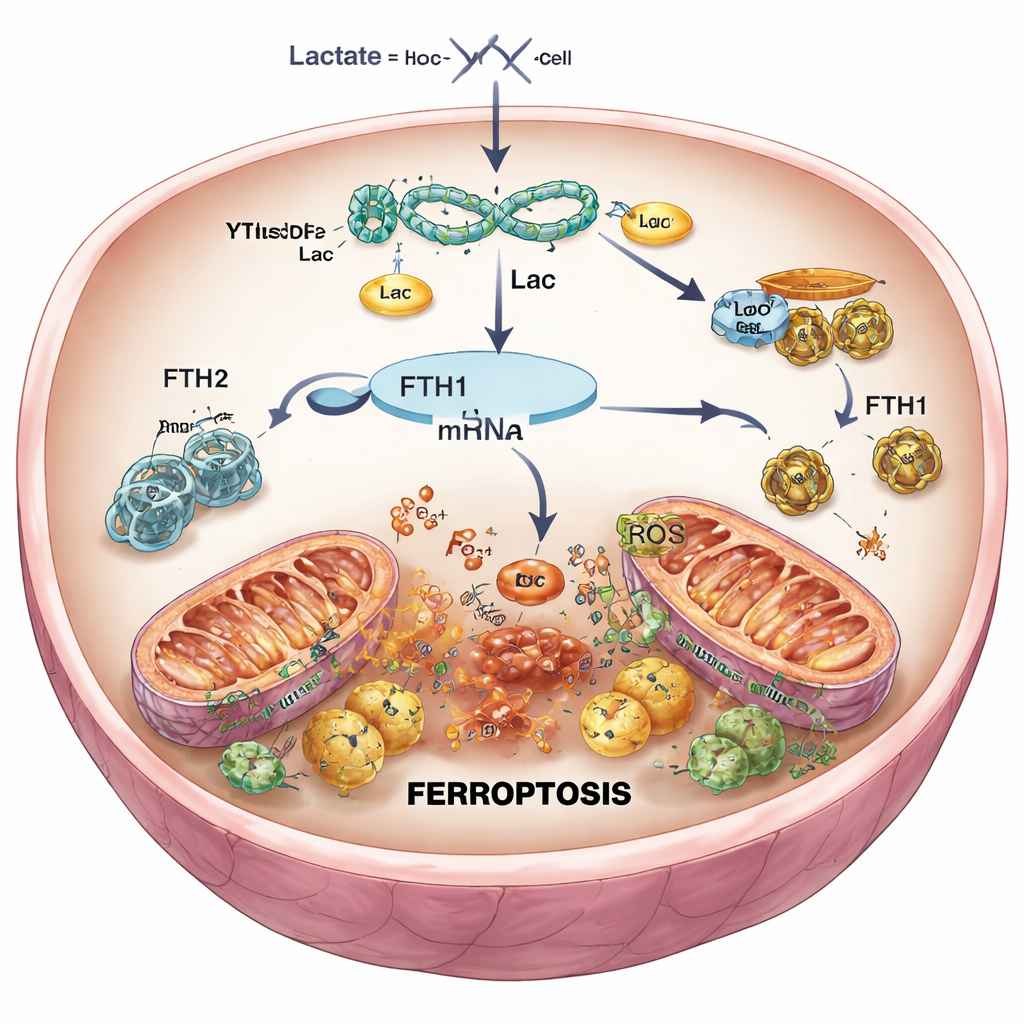
Przygaszanie sygnału śmierci w komórkach serca
Aby sprawdzić związek przyczynowo‑skutkowy, badacze użyli narzędzi genetycznych do selektywnego zmniejszenia YTHDF2 w kardiomiocytach i u myszy. Gdy YTHDF2 został stłumiony, poziomy FTH1 odbiły się, spadły żelazo i reaktywne formy tlenu, mitochondria utrzymały bardziej normalny kształt, a ogólne przeżycie komórek poprawiło się po symulowanej reperfuzji. U myszy mniejsze ilości YTHDF2 oznaczały mniejsze blizny po zawale i tkankę o zdrowszym wyglądzie. Jednak gdy równocześnie zmniejszono FTH1, korzyści te w dużej mierze zniknęły: żelazo ponownie wzrosło, powróciły uszkodzenia oksydacyjne, a rozmiar zawału się powiększył. Potwierdziło to, że YTHDF2 promuje ferroptozę głównie poprzez tłumienie FTH1, poluzowując kontrolę nad żelazem w komórkach serca.
Co to znaczy dla przyszłych terapii sercowych
Składając elementy razem, badanie opisuje nowy łańcuch zdarzeń: zatkane, a potem ponownie otwarte naczynie powoduje nagromadzenie kwasu mlekowego; kwas mlekowy zwiększa YTHDF2 przez laktylację; YTHDF2 niszczy instrukcje RNA dla białka strzegącego żelaza FTH1; a wynikające z tego przeciążenie żelazem wywołuje ferroptozę, pogłębiając uszkodzenie serca. Dla pacjentów przekaz jest optymistyczny: ta ścieżka oferuje kilka nowych punktów interwencji. Leki ograniczające szkodliwe sygnalizowanie kwasu mlekowego, blokujące specyficzną modyfikację YTHDF2 lub zachowujące funkcję FTH1 mogłyby uczynić reperfuzję w stanach nagłych bezpieczniejszą i ochronić więcej mięśnia sercowego. Choć wyniki te wymagają jeszcze potwierdzenia w tkankach ludzkich, otwierają obiecującą drogę do łagodniejszego i skuteczniejszego leczenia osób po zawale.
Cytowanie: Xiang, Z., Xiang, B., Ouyang, T. et al. Lactate regulates the YTHDF2-FTH1 axis to promote cardiomyocyte ferroptosis and aggravate myocardial ischemia-reperfusion injury. Sci Rep 16, 4865 (2026). https://doi.org/10.1038/s41598-026-35130-3
Słowa kluczowe: atak serca, kwas mlekowy, żelazozależna śmierć komórkowa, uszkodzenie niedokrwienie–reperfuzja, ochrona kardiomiocytów