Clear Sky Science · pl
Minimalny wirtualny zestaw danych do odtwarzalnego de novo montażu genomu triploidalnego
Dlaczego genom z trzema kopiami ma znaczenie
Wiele upraw i innych organizmów nie ma tylko dwóch kopii każdego chromosomu, jak u ludzi — mogą mieć trzy lub więcej. Odtworzenie tych dodatkowych kopii z danych sekwencjonowania DNA jest zaskakująco trudne, ponieważ kopie są bardzo podobne, lecz nie całkowicie identyczne. Ten artykuł przedstawia niewielki, ale starannie zaprojektowany „wirtualny” zestaw danych, który pozwala badaczom testować i porównywać oprogramowanie do montażu genomów na realistycznym problemie z trzema kopiami (triploidalnym), w warunkach całkowicie znanych i odtwarzalnych.
Budowanie prostego genomu zastępczego
Zamiast zaczynać od prawdziwej rośliny czy zwierzęcia, autor najpierw tworzy losowy odcinek DNA o długości jednego miliona nukleotydów, który służy jako czysty szablon. Ten szablon jest następnie skopiowany do trzech oddzielnych wersji, zastępując trzy zestawy chromosomów w organizmie triploidalnym. Aby naśladować, jak prawdziwe genomy powoli się zmieniają w czasie, w badaniu wprowadza się stałą liczbę drobnych zmian — podstawień pojedynczych liter — stopniowo do każdej kopii. Powtarzanie tego procesu przez 100 kroków daje trójki genomów o zróżnicowaniu od niemal identycznych do wyraźnie, lecz umiarkowanie różnych. Ten kontrolowany „gradient dywergencji” stanowi kręgosłup benchmarku.
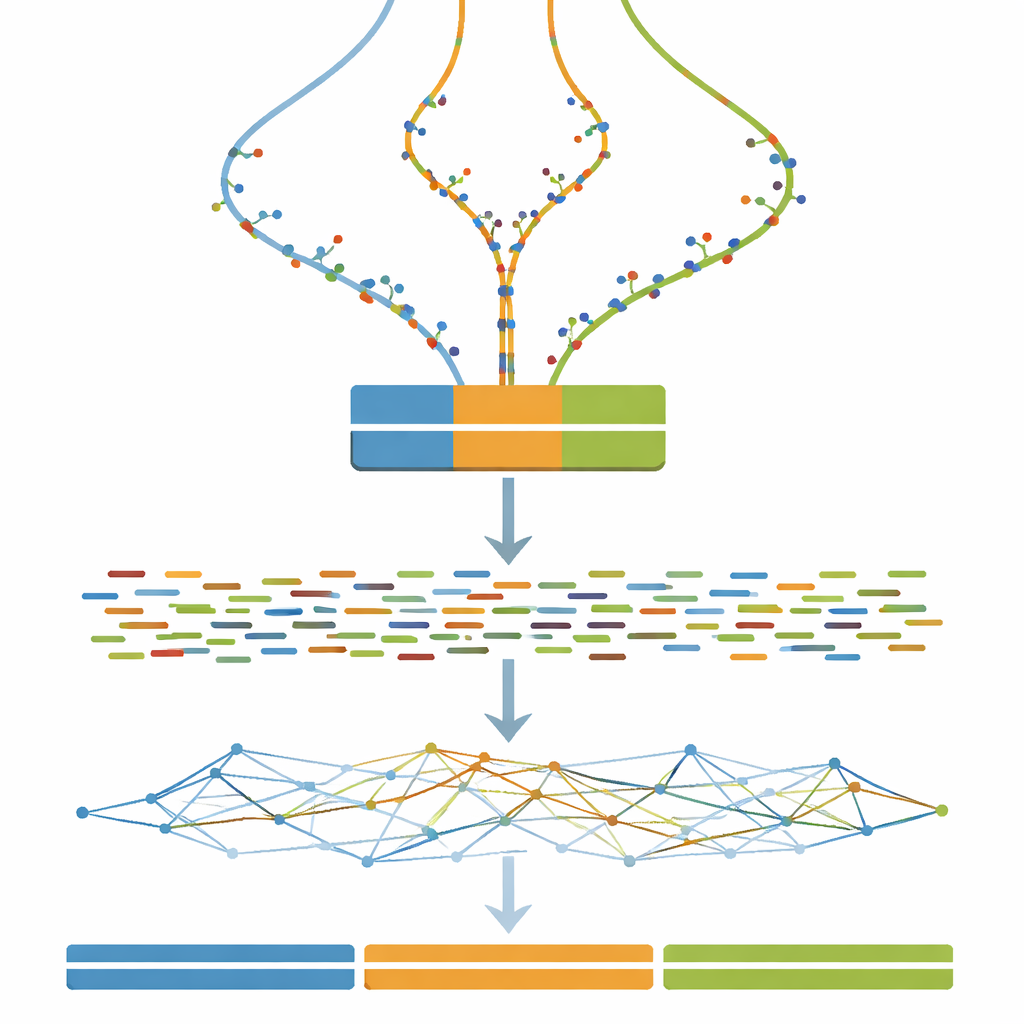
Przekształcanie wirtualnych genomów w wirtualne eksperymenty
Gdy każdy trzykrotny genom jest zdefiniowany, kolejnym krokiem jest naśladowanie tego, co zobaczyłoby urządzenie do sekwencjonowania DNA. Badanie używa szeroko stosowanego oprogramowania do symulacji krótkich sparowanych fragmentów DNA, podobnych do tych generowanych przez sekwenator Illumina, przy stałej i dość dużej głębokości pokrycia. Opcjonalne kroki oczyszczania naśladują typowe praktyki rzeczywiste, takie jak korekcja przypadkowych błędów sekwencjonowania i łączenie zachodzących na siebie par odczytów. W efekcie każdy, kto użyje zestawu danych, może testować nie tylko algorytmy montażu, lecz także wpływ typowych wyborów wstępnego przetwarzania na końcowe zmontowane genomy.
Testowanie strategii montażu pod obciążeniem
Istota pracy to olbrzymi eksperyment, w którym wszystkie symulowane odczyty są podawane do jednego programu do montażu genomu, zmieniając przy tym tylko jeden kluczowy parametr: rozmiar k-meru, czyli parametr kontrolujący, jak drobno oprogramowanie „dzieli” odczyty podczas rekonstrukcji genomu. Dla każdej kombinacji poziomu dywergencji (od 0 do 100 kroków) i rozmiaru k-meru (szeroki zakres nieparzystych wartości) budowany jest nowy montaż. Towarzyszące narzędzie ewaluacyjne mierzy potem ciągłość zmontowanych fragmentów, liczbę kawałków oraz to, jak ściśle ich łączna długość odpowiada znanej trzy-milionowej prawdzie. Wyniki podsumowywane są jako mapy cieplne, które ujawniają rozległe strefy, w których montaże łączy różne kopie w jedną, rozpada się na wiele drobnych fragmentów lub zbliża się do ideału trzech długich, dokładnych kontigów.
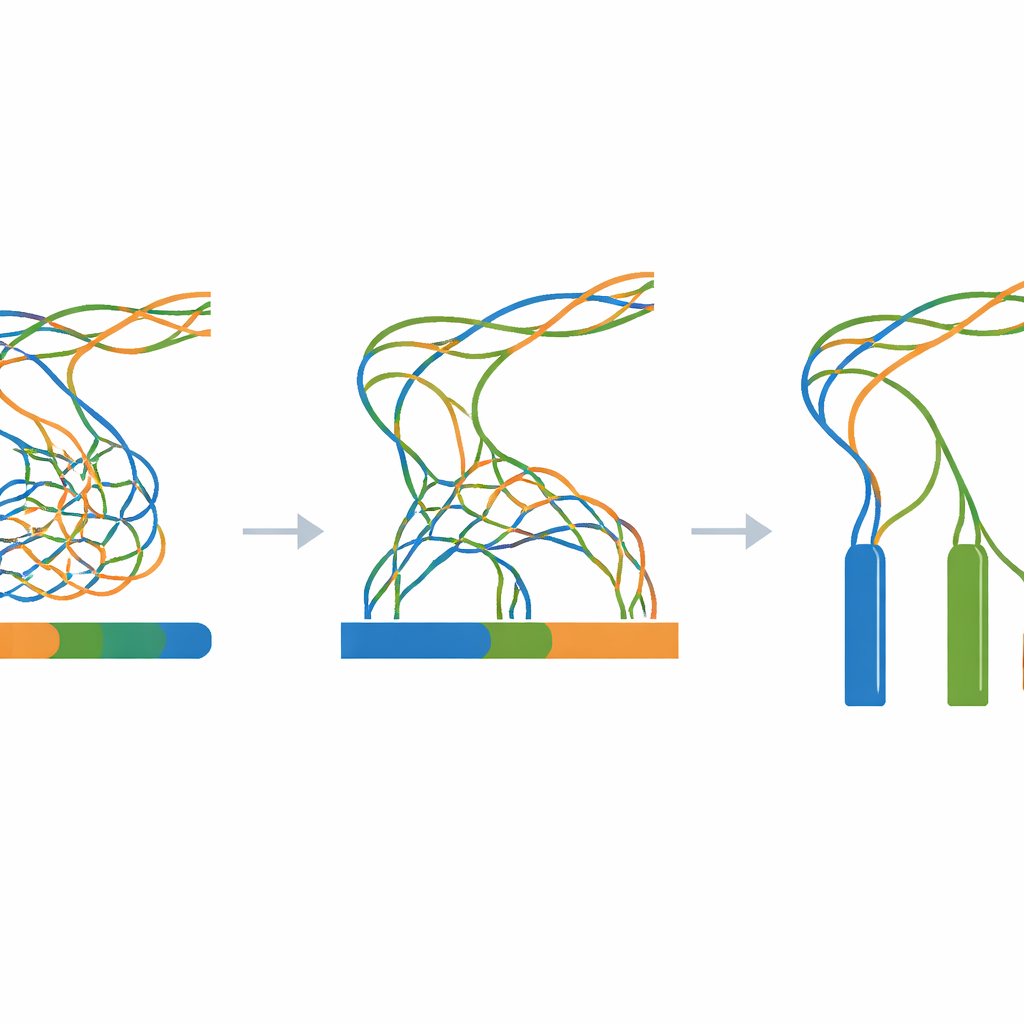
Przejrzyste odniesienie dla trudnych genomów
Ponieważ każdy etap jest syntetyczny i skryptowany — od początkowego losowego szablonu po końcowe montaże — badacze mogą odtworzyć cały tok pracy na dowolnym standardowym komputerze z Linuxem, używając tylko narzędzi open-source. Archiwum Zenodo powiązane w pracy zawiera genom szablonowy, wszystkie pośrednie zmienione sekwencje, wszystkie symulowane odczyty i każdy wynik montażu, wraz z logami i prostymi skryptami pomocniczymi. Kontrole techniczne potwierdzają, że proces mutacji zachowuje się zgodnie z oczekiwaniami, że symulowane odczyty odpowiadają żądanym długościom i pokryciu oraz że montaże pokazują przewidywany wzorzec: silne nadmierne łączenie, gdy trzy kopie są niemal identyczne, i wyraźniejsze rozdzielenie wraz z ich dalszym różnicowaniem.
Co to oznacza prostymi słowami
Mówiąc prosto, artykuł oferuje kontrolowany tor testowy dla oprogramowania próbującego odbudować trzy podobne księgi instrukcji z kup fragmentów pomieszanych. Poprzez stopniowe zwiększanie różnic między trzema „księgami” oraz systematyczną zmianę kluczowego ustawienia w procesie rekonstrukcji, zestaw danych ułatwia zobaczenie, kiedy i jak obecne metody zawodzą lub odnoszą sukces. Deweloperzy mogą używać go do strojenia nowych algorytmów, a użytkownicy lepiej zrozumieją, które ustawienia działają najlepiej dla genomów triploidalnych. Chociaż samo DNA jest sztuczne, wnioski, które umożliwia — dotyczące łączenia, rozdzielenia i wpływu wyborów parametrów — mają bezpośrednie znaczenie dla rzeczywistych prób rozszyfrowania złożonych genomów wielu istotnych gatunków.
Cytowanie: Ootsuki, R. Minimum virtual dataset for reproducible triploid de novo genome assembly. Sci Data 13, 382 (2026). https://doi.org/10.1038/s41597-026-06779-2
Słowa kluczowe: montaż genomu triploidalnego, ocena poliploidii, syntetyczny zestaw DNA, montaż de novo, optymalizacja k-merów