Clear Sky Science · pl
Metabolizm witaminy B2 zwiększa stabilność FSP1, zapobiegając ferroptozie
Jak powszechna witamina pomaga komórkom zdecydować między życiem a śmiercią
Nasze komórki nieustannie balansują między przetrwaniem a samozniszczeniem. Jedna dramatyczna forma śmierci komórkowej, zwana ferroptozą, przyciągnęła uwagę, ponieważ potrafi selektywnie zabijać komórki nowotworowe oporne na inne terapie. Badanie to ujawnia, że codzienny składnik diety — witamina B2, czyli ryboflawina — subtelnie przesuwa tę równowagę, stabilizując kluczowe białko ochronne. Zrozumienie tego ukrytego związku między dietą, metabolizmem komórkowym a śmiercią komórek nowotworowych może pomóc naukowcom w projektowaniu mądrzejszych terapii i dopracowywaniu przyszłych zaleceń żywieniowych.
Szczególny rodzaj rdzy wewnątrz komórek
Ferroptoza to rodzaj śmierci komórkowej napędzany procesem chemicznym przypominającym rdzewienie metalu: niekontrolowaną oksydacją lipidów w błonach komórkowych. Gdy te tłuszcze ulegają rozkładowi, błony tracą integralność, a komórki pękają. Komórki mają kilka mechanizmów obronnych, by temu zapobiec. Jedną z głównych tarcz jest enzym GPX4, który wykorzystuje małą cząsteczkę — glutation — do neutralizacji szkodliwych nadtlenków lipidowych. Drugą, równoległą ochroną jest białko FSP1, które osadza się na błonach komórkowych i używa małych, tłuszczopodobnych cząsteczek do przechwytywania niszczycielskich rodników, zanim się rozprzestrzenią. Komórki nowotworowe często zwiększają ekspresję FSP1, by unikać ferroptozy, co czyni to białko atrakcyjnym celem dla nowych leków przeciwnowotworowych. Do tej pory jednak nie wiadomo było, jak komórki kontrolują, ile FSP1 jest wytwarzane i jak długo ono przetrwa.
Budowanie komórkowego „wskaźnika paliwa” dla blokera śmierci
Aby odkryć ukrytych zarządców FSP1, badacze najpierw zaprojektowali ludzkie komórki kostne z reporterem fluorescencyjnym. Oznakowali naturalne białko FSP1 sygnałem zielonym i powiązali go z niebieskim sygnałem, który informuje o poziomie transkryptu FSP1. To sprytne rozwiązanie pozwoliło im odróżnić zmiany aktywności genu (niebieski) od zmian stabilności białka (zielony). Dysponując tym systemem dwóch kolorów, użyli CRISPR–Cas9 do systematycznego uszkadzania niemal każdego genu w genomie, a następnie sortowali komórki o wysokim lub niskim poziomie FSP1. Porównując, które guide RNA były wzbogacone w każdej grupie, zmapowali setki genów, które albo zwiększają, albo zmniejszają poziom FSP1, działając na poziomie kontroli genu lub obrotu białkowego.
Ukryta rola witaminy B2: tworzenie stabilizującego uchwytu
Wśród najbardziej uderzających trafień znalazły się dwa enzymy — kinaza ryboflawiny (RFK) i syntaza FAD (FLAD1), które przekształcają witaminę B2 w kofaktor zwany FAD. FSP1 jest białkiem flawoenzymatycznym, które normalnie mocno wiąże FAD, by przeprowadzać swoje reakcje chemiczne. Gdy RFK lub FLAD1 zostały usunięte, albo gdy komórki hodowano w medium pozbawionym witaminy B2, poziomy białka FSP1 gwałtownie spadały, mimo że aktywność jego genu pozostawała zbliżona. Zespół wykazał, że ta utrata czyni komórki znacznie bardziej podatnymi na ferroptozę przy blokadzie GPX4. Co ważne, sama witamina B2 nie działała jak klasyczny przeciwutleniacz: w czułym teście in vitro nie zatrzymała utleniania lipidów, w przeciwieństwie do witaminy E. Zamiast tego dodanie FAD (a częściowo jego prekursora FMN) do komórek z niedoborem przywracało zarówno poziomy FSP1, jak i odporność na śmierć ferroptotyczną, podczas gdy dodatkowa sama witamina B2 nie pomagała, jeśli brakowało enzymów przetwarzających.
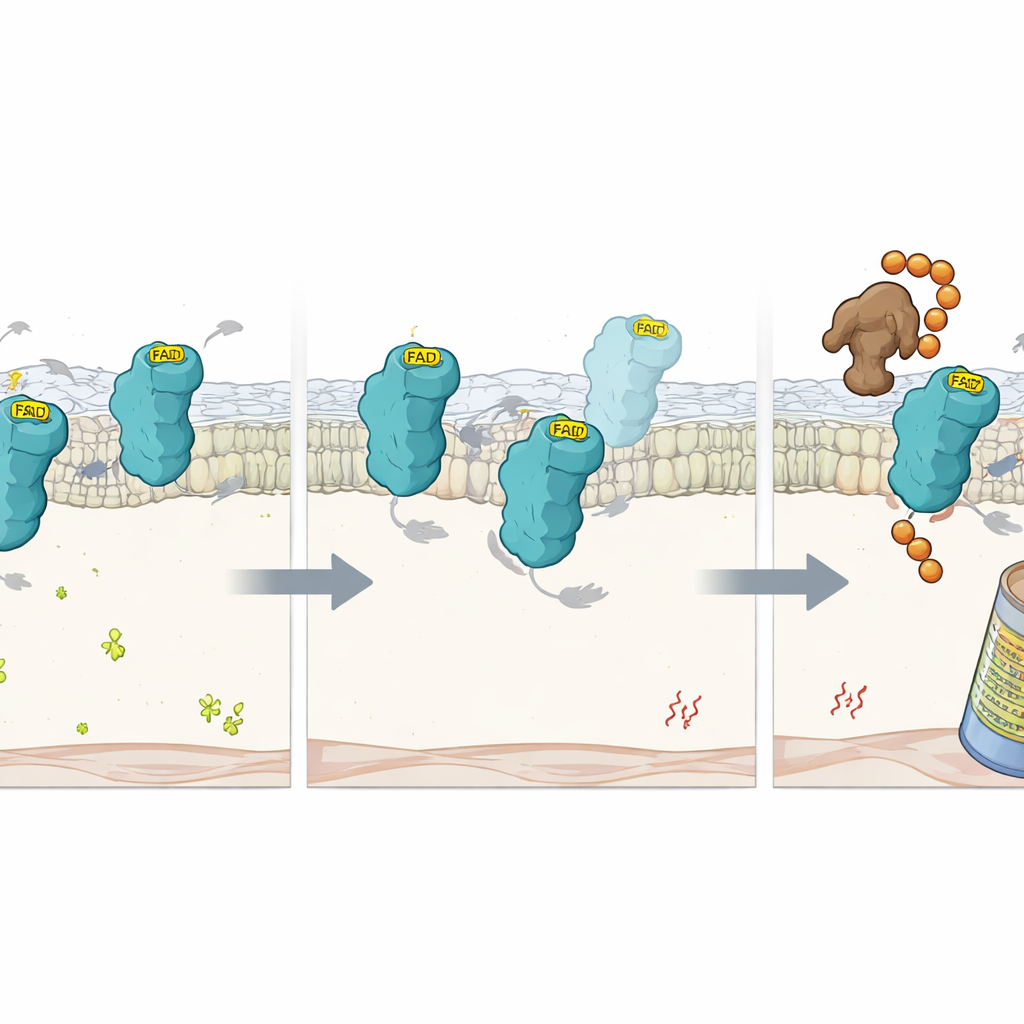
Co się dzieje, gdy brakuje kofaktora
Aby przyjrzeć się bliżej, naukowcy zbadali precyzyjne mutacje w FSP1, które uniemożliwiają mu wiązanie FAD. Te zmutowane białka nadal fałdowały się w zasadzie prawidłowo, ale utraciły FAD i aktywność katalityczną. W komórkach rozkładały się dużo szybciej niż normalne FSP1, chyba że zablokowano proteasom — komórkowy mechanizm rozdrabniania białek. To sugerowało, że samo wiązanie FAD działa jak stabilizujący uchwyt, chroniący FSP1 przed oznaczeniem jako wadliwe. W kolejnym ukierunkowanym ekranie CRISPR w warunkach niskiego FAD zespół zidentyfikował ligazę E3 nazwaną RNF8 jako kluczowy czynnik rozpoznający FSP1 pozbawione FAD. Gdy RFK został usunięty, RNF8 przyłączał łańcuchy ubikwityny do „pustego” białka, kierując je na zniszczenie przez proteasom. Usunięcie RNF8 spowolniło obrót FSP1 w komórkach ubogich w FAD, chociaż bez kofaktora nie przywróciło jego utraconej funkcji ochronnej.
Od molekularnych obwodów do pomysłów na terapię przeciwnowotworową
Składając te elementy w całość, autorzy proponują prosty, lecz silny model. Witamina B2, po przekształceniu w FAD przez RFK i FLAD1, wiąże się z FSP1 i jest niezbędna zarówno dla jego aktywności biochemicznej, jak i długowieczności. Gdy dostawy witaminy B2 lub jej przetwarzanie zawodzą, nowo wyprodukowany FSP1 nie może zabezpieczyć FAD, jest rozpoznawany przez RNF8 i szybko rozkładany przez proteasom, co pozostawia komórki bardziej narażone na uszkodzenia ferroptotyczne. Dane z nowotworów sugerują, że guzy o wyższej ekspresji RFK są bardziej oporne na leki indukujące ferroptozę, co podkreśla znaczenie tej ścieżki w rzeczywistych warunkach. Dla osób niebędących specjalistami kluczowy przekaz brzmi: dobrze znana witamina robi znacznie więcej niż działać jako prosty przeciwutleniacz — pomaga zadecydować, czy potężne białko-antyśmierć stoi na straży, czy zostaje usunięte. Poprzez regulowanie metabolizmu witaminy B2 lub stabilności FSP1, przyszłe terapie mogą lepiej wykorzystać ferroptozę do eliminacji komórek nowotworowych przy jednoczesnym oszczędzaniu tkanek zdrowych.
Cytowanie: Deol, K.K., Harris, C.A., Tomlinson, S.J. et al. Vitamin B2 metabolism promotes FSP1 stability to prevent ferroptosis. Nat Struct Mol Biol 33, 525–536 (2026). https://doi.org/10.1038/s41594-026-01759-x
Słowa kluczowe: ferroptoza, witamina B2, FSP1, śmierć komórek nowotworowych, metabolizm komórkowy