Clear Sky Science · pl
AF2BIND: przewidywanie miejsc wiązania małych cząsteczek przy użyciu reprezentacji par AlphaFold2
Znajdowanie celów leków w morzu białek
Współczesne leki często działają, przyczepiając się do drobnych szczelin i zagłębień na powierzchni białek wewnątrz naszych komórek. Nawet przy dzisiejszych ogromnych katalogach struktur białkowych wciąż zaskakująco trudno jest wcześniej określić, gdzie mała cząsteczka — potencjalny lek — rzeczywiście może się przyczepić. W tym badaniu wprowadzono AF2BIND, prosty, lecz potężny narzędzie obliczeniowe, które wydobywa informacje z wewnętrznych warstw AlphaFold2, przełomowego predyktora struktur białek, aby wskazać prawdopodobne miejsca wiązania leków w tysiącach ludzkich białek. Jego celem jest zawężenie poszukiwań nowych leków i ujawnienie ukrytych gorących punktów funkcjonalnych, które tradycyjne metody pomijają.
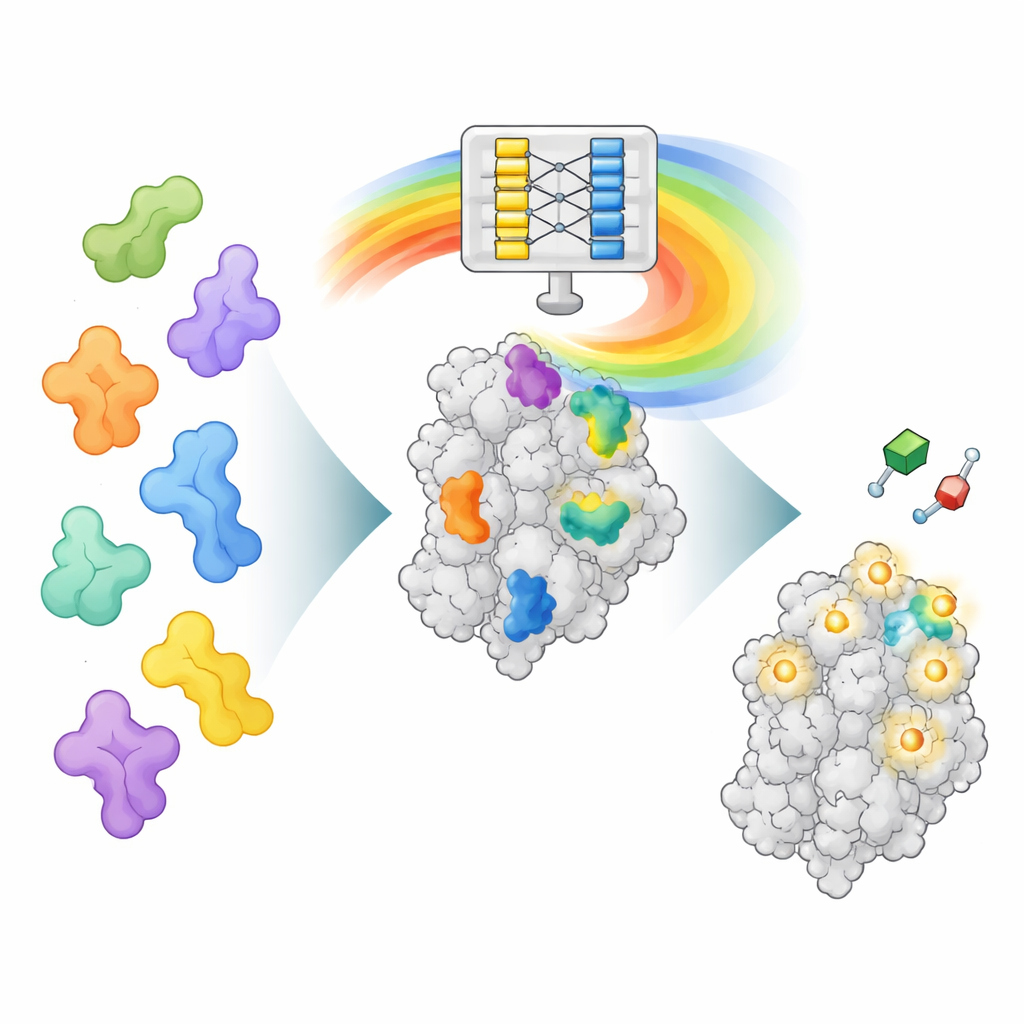
Nowy sposób odczytywania „umysłu” AlphaFold
AlphaFold2 był trenowany do przewidywania, jak łańcuch aminokwasów składa się w trójwymiarowe białko, a nie do znajdowania miejsc wiązania leków. Jednak w procesie uczenia się składania białek poznał też bogate wzorce dotyczące tego, jak różne części białek ze sobą oddziałują. AF2BIND sięga do jednej z tych wewnętrznych warstw danych, zwanej reprezentacją parową, która koduje, jak każda para pozycji aminokwasowych odnosi się w przestrzeni. Autorzy podają AlphaFold2 sekwencję białka wraz z jego szkieletem (backbone) i dodatkowo dołączają 20 ekstra aminokwasów, po jednym z każdego typu, jako osobne łańcuchy „przynęty”. AlphaFold2 oblicza wtedy, jak białko oddziałuje z każdą resztą przynęty. Te wzorce oddziaływań stają się danymi wejściowymi dla bardzo prostej regresji logistycznej, która ocenia dla każdej pozycji w białku prawdopodobieństwo, że należy do miejsca wiązania małej cząsteczki.
Przekształcanie ukrytych sygnałów w praktyczne przewidywania
Trenowanie AF2BIND wymagało starannie dobranego zestawu około 1900 struktur białko–ligand, w których małe cząsteczki były związane na podstawie wysokiej jakości dowodów eksperymentalnych. Badacze dołożyli znacznych starań, aby uniknąć „oszustwa” przez podobieństwo: podzielili dane tak, by białka testowe nie dzieliły ogólnego fałdu, sekwencji ani nawet kształtu kieszeni wiążącej z tymi użytymi do treningu. Na tym rygorystycznym zestawie referencyjnym reprezentacja par AF2 przewyższyła kilka alternatywnych osadzeń sieci neuronowych, w tym te oparte jedynie na sekwencji lub na projektowaniu sekwencji warunkowanym strukturą. Używając wyłącznie cech parowych, AF2BIND odzyskał około dwóch trzecich znanych reszt wiążących w najwyżej ocenianych przewidywaniach i wykazał silne wyniki we standardowych miarach klasyfikacji, pozostając przy tym odporny na umiarkowane zmiany kształtu białka i orientacji łańcuchów bocznych.
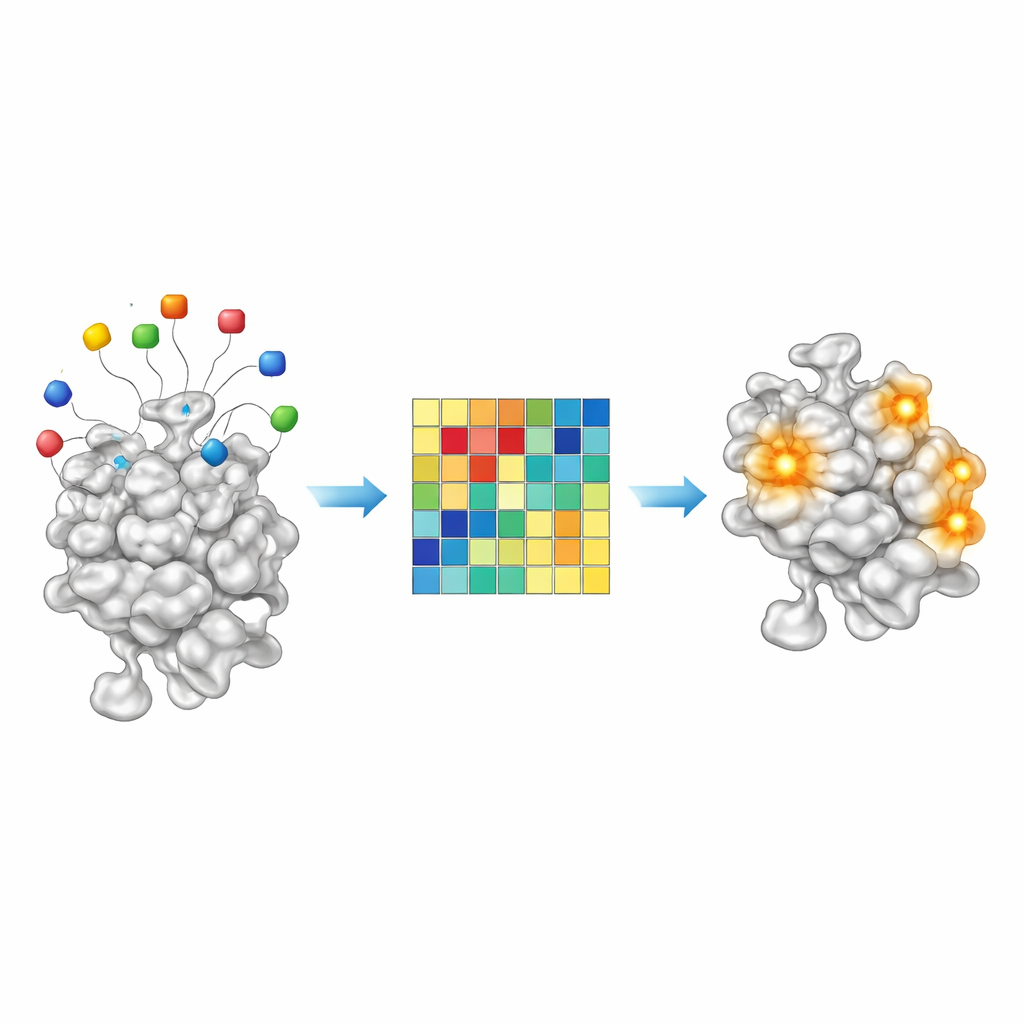
Odczytywanie chemicznych wskazówek z reszt przynęty
Ponieważ AF2BIND jest prostym modelem liniowym, jego decyzje są nadzwyczaj przejrzyste jak na współczesny system AI. Każdy z 20 aminokwasów przynęty wnosi mierzalny wkład do końcowego wyniku wiązania na danej pozycji białka. Analizując te wkłady w prawie 2000 kompleksach białko–ligand, autorzy odkryli, że niektóre kombinacje przynęt silniej reagują na tłuste, bogate w węgiel ligandy, podczas gdy inne aktywują się dla bardziej polarnego, hydrofilowego rodzaju cząsteczek. Innymi słowy, wzór aktywacji przynęt działa jak prymitywny chemiczny odcisk palca wskazujący, jakiego typu małe cząsteczki preferuje dana kieszeń. Sugeruje to, że w przyszłości podejścia podobne do AF2BIND mogą nie tylko wskazywać, gdzie lek mógłby się związać, ale też sugerować rodzaj chemii najlepiej pasującej do danej kieszeni.
Skanowanie proteomu ludzkiego w poszukiwaniu nowych kieszeni
Wyposażeni w wytrenowany model, zespół uruchomił następnie AF2BIND na strukturach całego proteomu ludzkiego przewidzianych przez AlphaFold. Po odcięciu obszarów o niskim zaufaniu i podzieleniu bardzo dużych białek na nadające się do analizy fragmenty strukturalne, sklasteryzowali pobliskie reszty o wysokich wynikach w kandydackie miejsca wiążące. AF2BIND przewidział ponad 20 000 takich miejsc w więcej niż 13 000 białek. Co zaskakujące, większość z nich nie pokrywała się z kieszeniami wywnioskowanymi metodami opartymi na homologii, takimi jak AlphaFill, które kopiują ligandy z pokrewnych struktur krystalograficznych, ani z powszechnie używanym narzędziem do znajdowania kieszeni P2Rank. Wiele miejsc wykrywanych tylko przez AF2BIND jest płytszych lub bardziej rozproszonych niż klasyczne zatopione kieszenie i często zbiega się z regionami wiążącymi peptydy, RNA, DNA lub inne białka — interfejsami, które mimo to mogą być podatne na działanie małych cząsteczek.
Implikacje dla odkrywania leków i chorób
Aby ocenić, jak obiecujące mogą być te nowo zasugerowane miejsca dla projektowania leków, autorzy użyli niezależnego narzędzia oceniającego „druggability” na podstawie wielkości kieszeni, zamknięcia i środowiska chemicznego. Średnio miejsca wskazane przez AF2BIND uzyskały wyniki powyżej powszechnie stosowanego progu dla atrakcyjnych celów lekowych, w tym tych znalezionych w białkach powiązanych z chorobami dziedzicznymi. Po zestawieniu z eksperymentami chemoproteomicznymi, które znakują reaktywne cysteiny w komórkach, AF2BIND i P2Rank razem wyjaśniły prawie połowę obserwowanych rejonów podatnych na ligandację, każda metoda wykrywając przypadki, które druga pominęła. Praca ta pokazuje, że wewnętrzne reprezentacje uczone przez sieci predykcji struktur można wykorzystać do mapowania prawdopodobnych miejsc wiązania leków na dużą skalę, bez wcześniejszej wiedzy o konkretnym ligandzie. Dla laików kluczowa wiadomość jest taka, że te same przełomowe osiągnięcia AI, które przewidują kształty białek, zaczynają ujawniać, gdzie i jak leki mogłyby najlepiej uchwycić te kształty, potencjalnie przyspieszając poszukiwania nowych terapii i oświetlając wcześniej ukryte punkty kontroli w naszych białkach.
Cytowanie: Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods 23, 626–635 (2026). https://doi.org/10.1038/s41592-026-03011-2
Słowa kluczowe: miejsca wiązania białek, poszukiwanie leków, AlphaFold2, biologia obliczeniowa, bioinformatyka strukturalna