Clear Sky Science · pl
DECODE: oparte na głębokim uczeniu uniwersalne ramy dekonwolucji dla różnych danych omicznych
Dlaczego te badania są ważne
Współczesna biomedycyna jest zalana pomiarami naszych tkanek: które geny są aktywne, które białka występują i jakie małe cząsteczki zasilają komórki. Jednak większość tych pomiarów pochodzi z mieszanek próbek, gdzie wiele typów komórek jest zmieszanych. Badanie stojące za DECODE przedstawia potężne ramy sztucznej inteligencji, które potrafią rozdzielić te sygnały, określając, które komórki i stany komórkowe są obecne, nawet w bardzo różnych rodzajach danych. Ta zdolność może przyspieszyć badania nad rakiem, odpornością i chorobami metabolicznymi, a także lepiej wykorzystać istniejące próbki z biobanków.
Wgląd w zmieszane tkanki
Każdy narząd to wspólnota różnych typów komórek — komórek układu odpornościowego, komórek strukturalnych, komórek macierzystych i innych. W zdrowiu i w chorobie często zmienia się nie tylko to, co robi każda komórka, lecz także ile jest każdego typu i w jakim są stanie. Technologie jednoukładowe potrafią mierzyć komórki indywidualnie, ale są kosztowne i technicznie wymagające, szczególnie w dużych kohortach pacjentów lub dla starych przechowywanych próbek. Z kolei konwencjonalne eksperymenty „bulk” mieszają tysiące lub miliony komórek i odczytują sygnał uśredniony. Algorytmy dekonwolucji starają się odwrócić to mieszanie: mając dane bulk i referencję jednoukładową, estymują proporcje poszczególnych typów komórek w tkance.
Ograniczenia narzędzi jednofunkcyjnych
Istniejące narzędzia dekonwolucyjne są przeważnie dostosowane do jednego rodzaju pomiaru, takiego jak aktywność genów (transkryptomika) czy białka (proteomika). Często zakładają specyficzne zachowania statystyczne, które nie sprawdzają się w innych typach danych, i mają trudności, gdy w tkance bulk pojawiają się typy komórek nieobecne w referencji. Silne efekty partii — różnice między dawcami, instrumentami lub stanami zdrowia — mogą dodatkowo rozmywać biologiczne sygnały. Co istotne, brakowało praktycznej metody dla metabolomiki, dziedziny badającej małe cząsteczki, które często są najbliżej objawów klinicznych. W rezultacie naukowcy analizujący kohorty multiomiczne musieli żonglować kilkoma wyspecjalizowanymi narzędziami, każde z własnymi niuansami, co utrudniało porównywanie wyników między badaniami i typami danych.
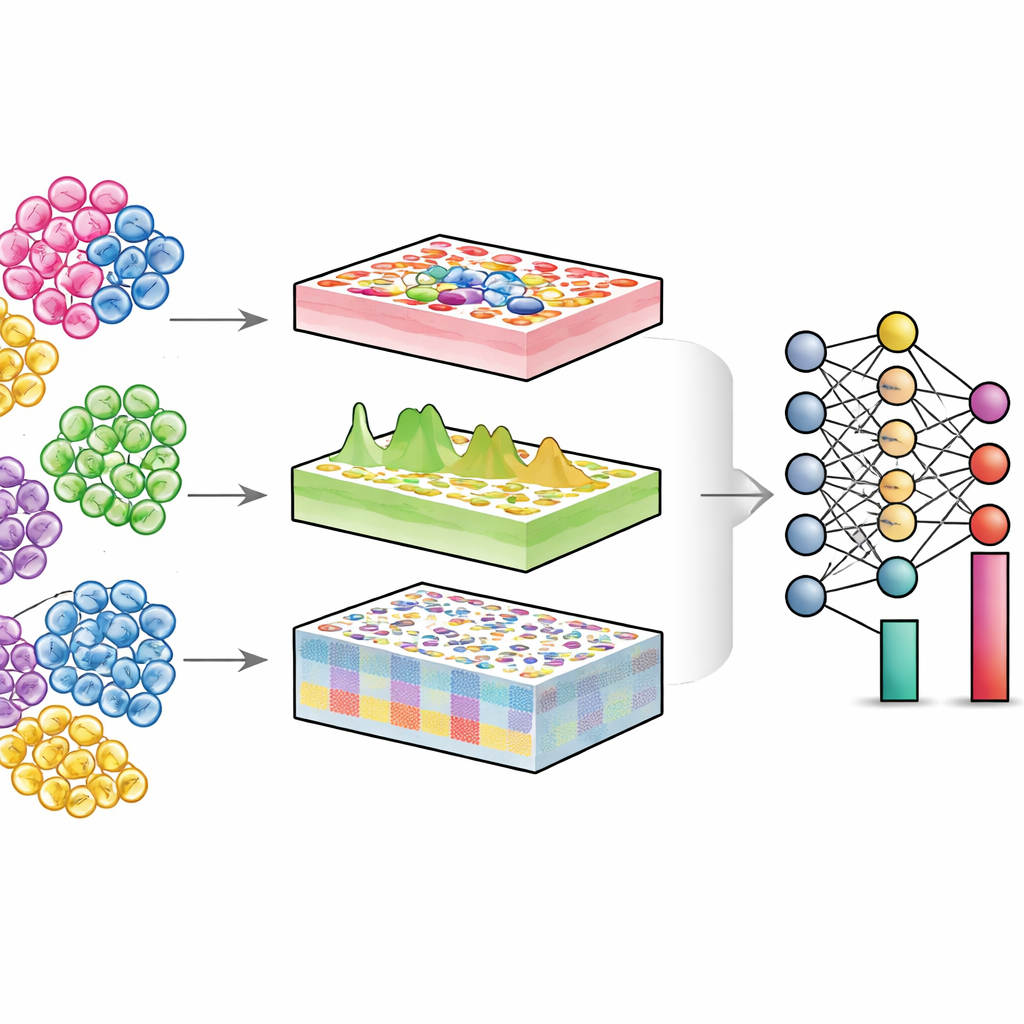
Uniwersalny silnik rozdzielający
DECODE rozwiązuje te wyzwania, traktując dekonwolucję jako elastyczny problem uczenia głębokiego, który może obsługiwać geny, białka i metabolity w ujednolicony sposób. Najpierw syntetyzuje „pseudotkanki” przez cyfrowe mieszanie profili jednoukładowych w losowych proporcjach, tworząc bogaty zbiór treningowy, w którym znana jest prawdziwa kompozycja komórkowa. Etap uczenia adwersarnego uczy następnie enkodera mapować zarówno tkanki rzeczywiste, jak i pseudotkanki do wspólnej reprezentacji, gdzie różnice techniczne są zredukowane, a biologicznie istotne wzorce zachowane. Kolejny moduł denoisingowy, prowadzony uczeniem kontrastowym, uczy się oddzielać prawdziwe sygnały tkankowe od sztucznego szumu. Ten etap sprawia, że DECODE jest odporny na brakujące typy komórek w danych referencyjnych i na błędy pomiarowe. Na koniec oczyszczone cechy trafiają do modułu dekonwolucji, który estymuje albo absolutne, albo względne obfitości typów i stanów komórkowych, w zależności od kompletności referencji.
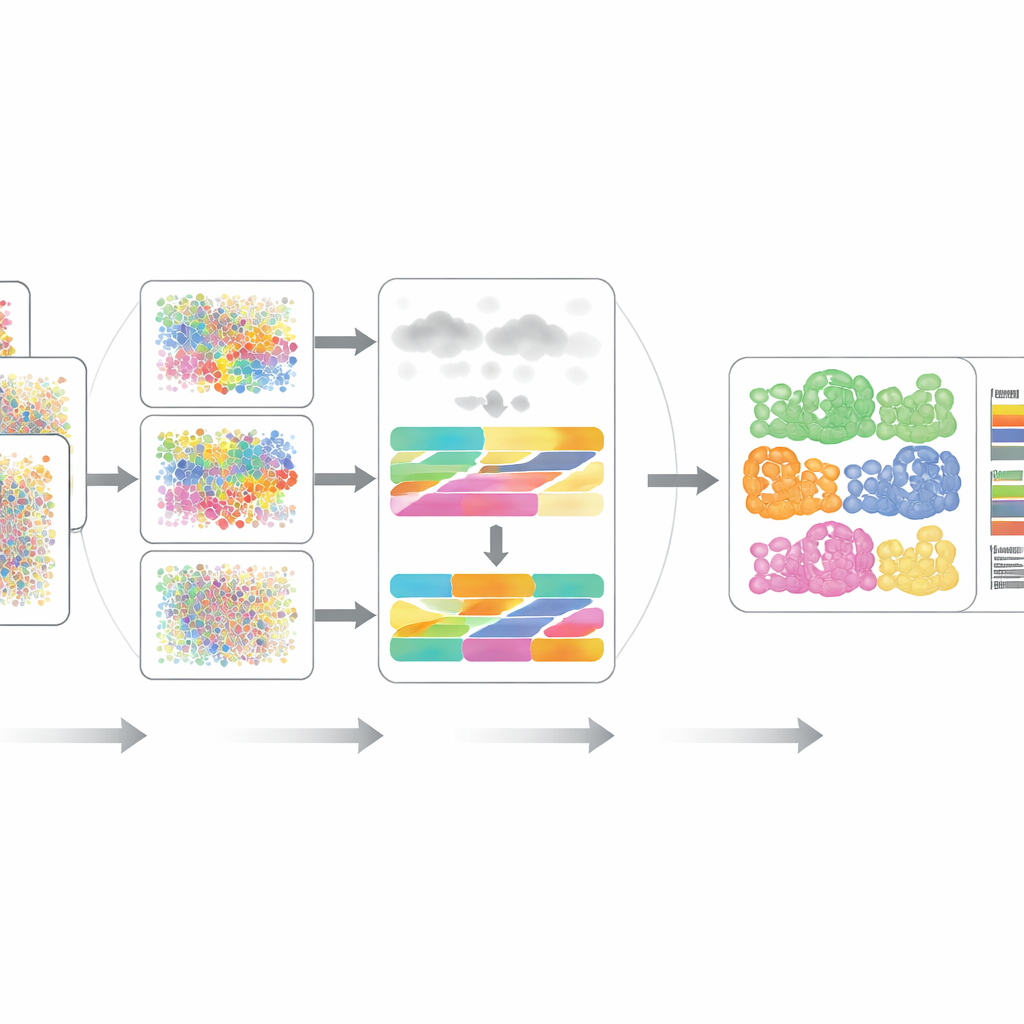
Testy działania DECODE
Autorzy rygorystycznie przetestowali DECODE na 15 zbiorach danych obejmujących siedem realistycznych scenariuszy, w tym różnych dawców, stanów chorobowych, warunków zdrowotnych, platform eksperymentalnych, a nawet pomiarów przestrzennych. W transkryptomice i proteomice DECODE zazwyczaj dorównywał lub przewyższał narzędzia będące stanem wiedzy pod względem dokładności, przy zachowaniu rozsądnego czasu obliczeń i użycia pamięci. Co kluczowe, DECODE był jedyną metodą dostarczającą wiarygodne wyniki dla danych metabolomicznych, gdzie występuje mniej cech, a różne typy komórek mogą wyglądać myląco podobnie. Ramy okazały się także sprawne w śledzeniu stanów komórkowych — takich jak postęp wzdłuż trajektorii rozwojowej, fazy cyklu komórkowego czy odpowiedzi na leczenie farmakologiczne — a nie tylko statycznych typów komórek.
Odporność na hałas i niekompletność danych z rzeczywistego świata
W prawdziwych tkankach często występują typy komórek nieuchwycone w referencjach jednoukładowych, a szum eksperymentalny może zniekształcić wiele cech jednocześnie. Badacze zasymulowali te problemy, dodając nieznane typy komórek oraz wprowadzając różne rodzaje szumu i braków danych w transkryptomice, proteomice i metabolomice. W większości ustawień DECODE pozostał najdokładniejszą metodą, a w metabolomice — jedyną, która się nie załamała. Pokazali też, że DECODE daje bardzo spójne odpowiedzi przy zastosowaniu dopasowanych pomiarów genów i białek z tych samych próbek komórek krwi, co jest kluczowe do porównywania zmian typów komórek między warstwami omicznymi w dużych kohortach.
Nowe biologiczne wnioski z kohort multiomicznych
Wyposażeni w to ujednolicone narzędzie, autorzy ponownie przeanalizowali złożone zbiory danych dotyczące chorób. W raku piersi połączyli kohorty transkryptomiczne i proteomiczne, aby pokazać, jak komórki odpornościowe i wspomagające komórki zrębowe zmieniają się między guzami niemetastatycznymi, przerzutującymi guzami pierwotnymi a przerzutami do mózgu. Wzorce takie jak większa liczba limfocytów T i komórek przypominających perivascular w zmianach niemetastatycznych oraz zwiększenie liczby komórek B w zaawansowanej chorobie zgadzają się z wcześniejszymi badaniami i je rozszerzają. W wątrobie mysiej DECODE zintegrował kohorty transkryptomiczne, proteomiczne i metabolomiczne, by śledzić, jak hepatocyty, komórki śródbłonka i komórki odpornościowe rezydujące zmieniają się w zależności od diety i modeli chorób wątroby, odtwarzając znane tendencje, takie jak wzrost ułamka komórek Kupffera w stanach zapalnych.
Co to oznacza na przyszłość
Dla czytelnika niebędącego specjalistą główne przesłanie jest takie, że DECODE działa jak inteligentna pryzma dla danych biomedycznych: mając zmieszane pomiary z tkanek, potrafi rozdzielić wkład wielu różnych typów i stanów komórkowych, i robi to wiarygodnie w kilku rodzajach odczytów molekularnych. Pozwala to naukowcom uzyskać znacznie więcej informacji z istniejących kohort multiomicznych i biobanków bez konieczności pozyskiwania nowych danych jednoukładowych dla każdego projektu. Choć metoda wciąż zależy od jakości i zakresu dostępnych referencji jednoukładowych, a zasoby metabolomiczne pozostają ograniczone, DECODE stanowi istotny krok w kierunku rutynowej interpretacji na poziomie komórkowym dużych badań ludzkich, z potencjalnymi korzyściami dla zrozumienia mechanizmów chorób i prowadzenia medycyny precyzyjnej.
Cytowanie: Zhao, T., Liu, R., Sun, Y. et al. DECODE: deep learning-based common deconvolution framework for various omics data. Nat Methods 23, 596–608 (2026). https://doi.org/10.1038/s41592-026-03007-y
Słowa kluczowe: dekonwolucja multiomiki, referencja jednoukładowa, głębokie uczenie w biologii, analiza metabolomiki, skład komórkowy