Clear Sky Science · pl
Zmiana szybkości mutacji i powtarzające się błędy sekwencyjne w filogenetyce na skalę pandemiczną
Dlaczego ma to znaczenie dla przyszłych epidemii
Gdy nowy wirus rozprzestrzenia się po świecie, naukowcy ścigają się z czasem, by odczytać jego kod genetyczny i odtworzyć drzewo rodowe. Takie drzewa pomagają śledzić, jak powstają warianty, jak szybko się rozprzestrzeniają i czy środki kontrolne działają. Jednak w trakcie COVID-19 laboratoria tak szybko zsekwencjonowały miliony genomów SARS‑CoV‑2, że ukryte błędy i narożne zjawiska w danych zaczęły zniekształcać obraz. Artykuł przedstawia nowe metody oczyszczania i interpretacji tak rozległych zestawów genetycznych, oferując jaśniejszy obraz tego, jak wirus pandemiczny rzeczywiście ewoluuje i przemieszcza się w populacjach.
Wyzwaniem jest sensowne zinterpretowanie milionów genomów
Epidemiologia genomowa przekształca genomy wirusów w praktyczne informacje dla decyzji zdrowia publicznego. Dla SARS‑CoV‑2 udostępniono na świecie ponad 20 milionów genomów. Tradycyjne narzędzia ewolucyjne powstały z myślą o skromniejszych problemach, takich jak porównywanie genów między gatunkami, a nie o obsłudze milionów niemal identycznych sekwencji wirusowych napływających w czasie rzeczywistym. Na taką skalę szczególnie kłopotliwe stają się dwa zjawiska. Po pierwsze, niektóre miejsca w genomie wirusa mutują znacznie częściej niż inne, co może sprawić, że niezwiązane ze sobą wirusy będą wyglądać podejrzanie podobnie. Po drugie, powtarzające się błędy techniczne w sekwencjonowaniu i przetwarzaniu danych mogą naśladować prawdziwe mutacje. Oba efekty generują „fałszywe echa” w drzewie ewolucyjnym, tworząc niepewność co do tego, którym gałęziom i grupowaniom można ufać.
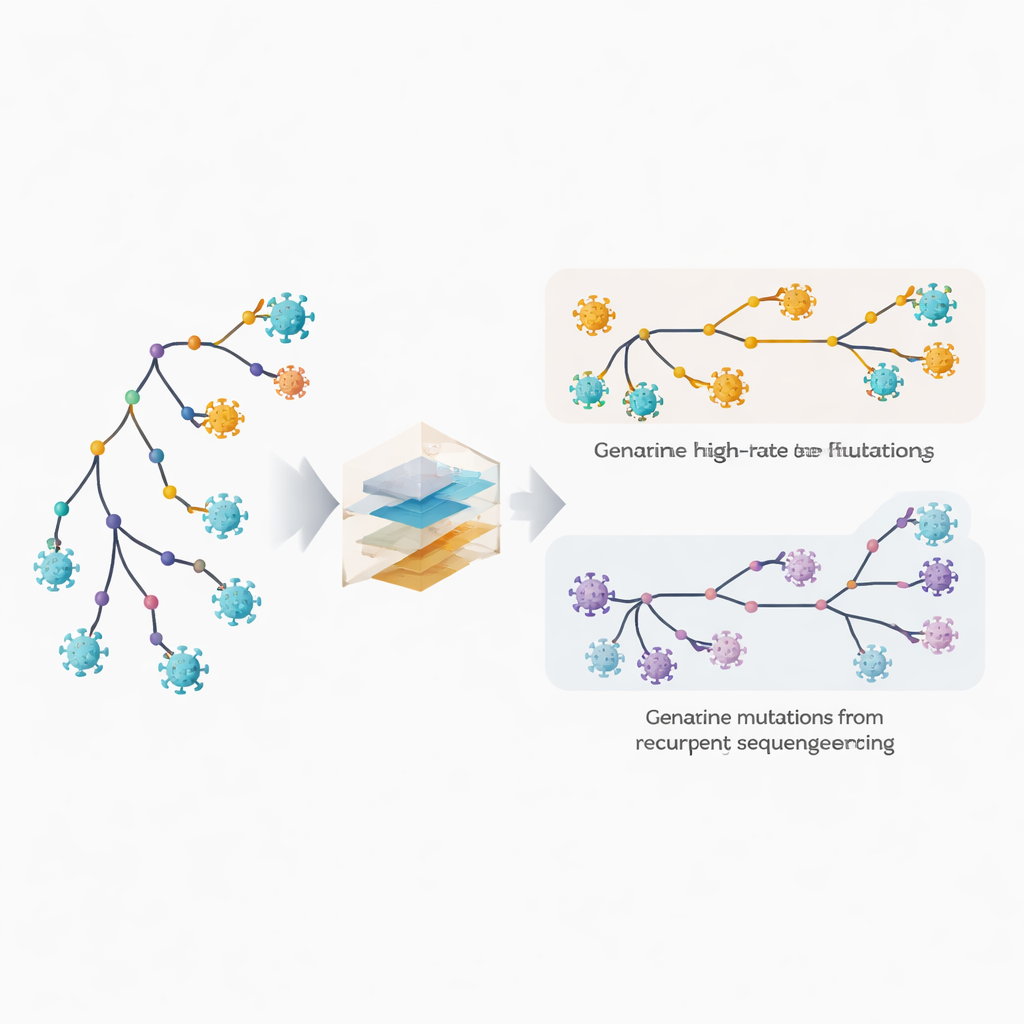
Wykrywanie szybko zmieniających się miejsc i ukrytych błędów
Autorzy rozszerzają swoje oprogramowanie filogenetyczne MAPLE o modele traktujące każdą pozycję w genomie wirusa jako mającą własne właściwości. Zamiast zakładać kilka przeciętnych szybkości mutacji, metoda estymuje oddzielną szybkość dla każdego miejsca, wykorzystując ogromną liczbę dostępnych genomów. Równocześnie pozwala każdemu miejscu mieć własne prawdopodobieństwo powtarzającego się błędu sekwencjonowania lub ustalania konsensusu. Kluczowy trik polega na porównaniu, jak często zmiana pojawia się na głębokich, wewnętrznych gałęziach drzewa, które odzwierciedlają starsze, wspólne zdarzenia, w stosunku do zewnętrznych końcówek, odpowiadających poszczególnym genomom. Prawdziwe mutacje biologiczne mają tendencję do występowania zarówno na gałęziach wewnętrznych, jak i terminalnych, podczas gdy błędy techniczne pojawiają się głównie na końcach. Wykorzystując ten wzorzec, metoda potrafi rozdzielić autentyczną szybką ewolucję od powtarzających się pomyłek.
Szybsze algorytmy dla zatłoczonego drzewa życia
Obsługa milionów genomów normalnie wymagałaby ogromnej mocy obliczeniowej. Aby analiza była praktyczna, zespół przeprojektował sposób, w jaki MAPLE przechowuje i aktualizuje informacje o sekwencjach na drzewie. Zamiast porównywać każdy genom z jednym stałym odniesieniem, oprogramowanie wybiera „lokalne punkty odniesienia” wewnątrz drzewa i zapisuje pobliskie genomy jako różnice względem tych kotwic. Ta zwarta reprezentacja przyspiesza porównania między odległymi częściami drzewa. Dodatkowe usprawnienia dopracowują sposób dodawania nowych próbek do istniejącego drzewa, strojenia długości gałęzi i badania prawdopodobieństwa alternatywnych kształtów drzewa, z opcjami uruchamiania najbardziej wymagających kroków równolegle na wielu rdzeniach procesora.
Testowanie metody i oczyszczanie danych rzeczywistych
Aby sprawdzić działanie swoich modeli, autorzy najpierw stworzyli realistyczne symulowane zestawy danych SARS‑CoV‑2 o znanych wzorcach mutacji i osadzonych błędach sekwencji. W tych testach nowe podejście odtworzyło bardziej wierne drzewa ewolucyjne i zlokalizowało indywidualne błędy z wysoką precyzją, zwłaszcza gdy uwzględniono dziesiątki tysięcy genomów lub więcej. Następnie przeszli do danych rzeczywistych, analizując miliony sekwencji SARS‑CoV‑2, dla których dostępne były surowe odczyty. Porównując dwa różne pipeline’y do budowy konsensusu, wyodrębnili konkretne pozycje w genomie wielokrotnie dotknięte artefaktami, takimi jak problemy z przyłączaniem starterów (primerów) czy wywoływanie konsensusu z uprzedzeniem względem referencji. Podejrzane miejsca zostały zamaskowane w dalszych analizach, a genomy wykazujące oznaki zanieczyszczenia lub mieszanej infekcji odfiltrowano, uzyskując opiekowany wyrównanie ponad dwóch milionów wysokiej jakości sekwencji.
Jaśniejszy globalny obraz drzewa rodowego wirusa
Korzystając z oczyszczonego zestawu danych, autorzy zrekonstruowali globalne drzewo filogenetyczne SARS‑CoV‑2 i zobrazowali, jak główne warianty odnoszą się do siebie. Ich drzewo czasem proponuje subtelnie inne relacje niż wcześniejsze publiczne drzewa, często w sposób wymagający mniej zdarzeń mutacyjnych i lepiej pasujący do modelu statystycznego. Ramy analizy również uwypuklają miejsca, gdzie etykiety linii mogą być niespójne z leżącą u ich podstaw historią genetyczną, sygnalizując możliwe rekombinanty lub problematyczne genomy do bliższej inspekcji. Chociaż pewne wyzwania pozostają — na przykład przeuczenie przy skąpych danych czy wpływ silnie zanieczyszczonych próbek — praca pokazuje, że teraz wykonalne jest budowanie bardziej wiarygodnych drzew ewolucyjnych na skalę pandemii. Dla czytelnika niebędącego specjalistą sedno jest takie: lepsze radzenie sobie z błędami i gorącymi punktami mutacji prowadzi do ostrzejszych wglądów w to, jak patogeny się rozprzestrzeniają i zmieniają, pomagając naukowcom i agencjom zdrowia szybciej i pewniej reagować przy przyszłych ogniskach choroby.
Cytowanie: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
Słowa kluczowe: genomika SARS-CoV-2, metody filogenetyczne, błędy sekwencjonowania, zmienność częstości mutacji, epidemiologia genomowa