Clear Sky Science · pl
Przenośne modele enantioselektywności z rzadkich danych
Inteligentniejszy sposób na znalezienie odpowiedniego katalizatora
Chemicy często poszukują lepszych leków i materiałów, próbując łączyć atomy węgla w bardzo określone układy trójwymiarowe. Uzyskanie subtelnego efektu „prawej” versus „lewej” formy — znanego jako enantioselektywność — zwykle wymaga testowania wielu metalicznych katalizatorów i warunków reakcji metodą prób i błędów. W artykule przedstawiono metodę wykorzystania stosunkowo niewielkiej ilości danych eksperymentalnych, połączonej z szybkimi obliczeniami komputerowymi, aby przewidzieć, które katalizatory na bazie niklu dadzą pożądaną konfigurację enancjomeryczną w szerokim zakresie reakcji, co może oszczędzić chemikom tygodnie lub miesiące pracy laboratoryjnej.
Dlaczego kontrolowanie „ręczności" cząsteczek jest tak trudne
Wiele leków i produktów naturalnych występuje w formach lustrzanych, które mogą zachowywać się bardzo różnie w organizmie. Katalizatory faworyzujące jedną formę nad drugą są zatem niezwykle cenne. Projektowanie takich katalizatorów jest jednak skomplikowane. Tradycyjna chemia kwantowa teoretycznie potrafi obliczyć, która ścieżka reakcji jest preferowana, ale drobne błędy energetyczne przekładają się na duże pomyłki w przewidywanej selektywności, a obliczenia są czasochłonne. Prostszе modele statystyczne z kolei są szybkie, lecz często pomijają szczegółowy „taniec” między metalicznym katalizatorem a reagującymi cząsteczkami, zwłaszcza gdy mechanizm reakcji może subtelnie zmieniać się w zależności od użytych partnerów.
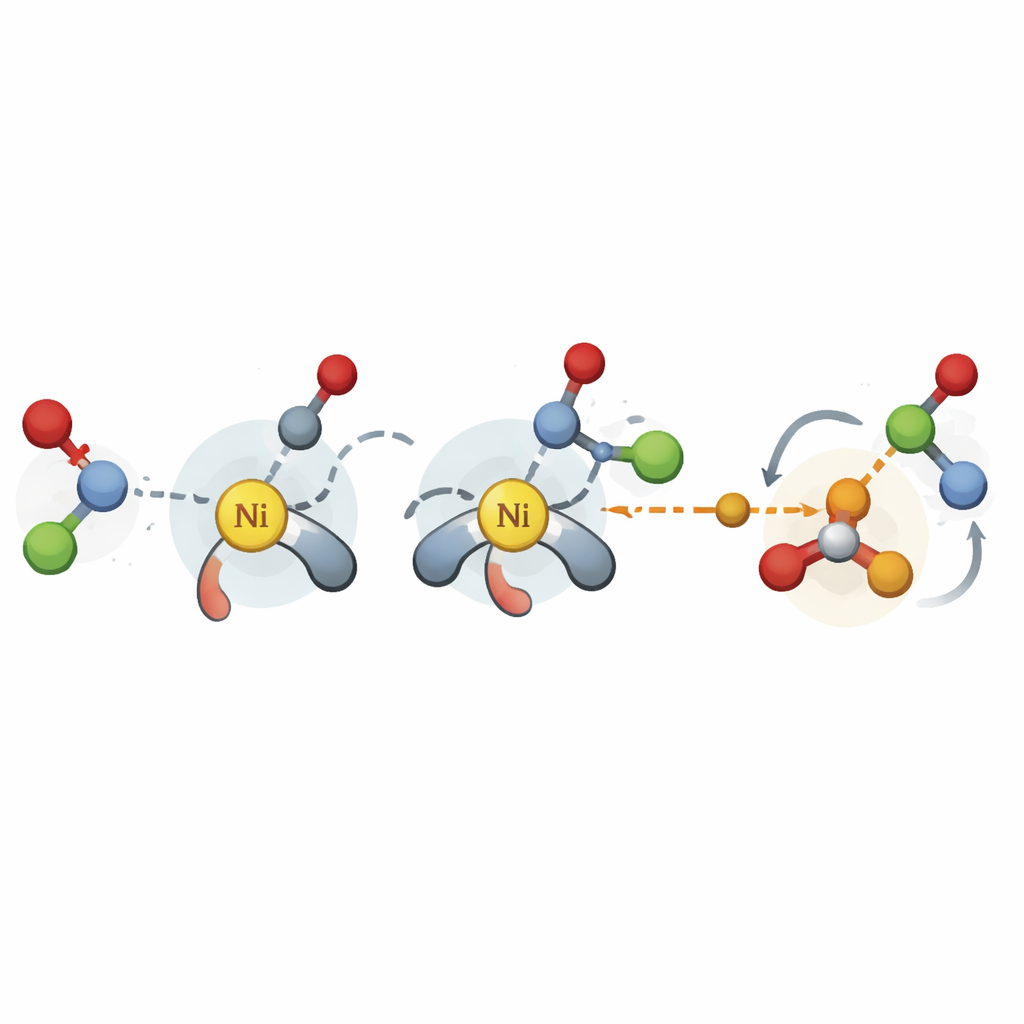
Uchwycenie kluczowych momentów reakcji
Autorzy wypełniają tę lukę, koncentrując się na najważniejszych etapach niklowo-katalizowanego sprzęgania krzyżowego: krokach, w których tworzone są nowe wiązania węgiel–węgiel oraz oddawaniu końcowego produktu. Zamiast wykonywać kosztowne, wysokopoziomowe symulacje, stosują uproszczoną metodę kwantową do generowania struktur trójwymiarowych kluczowych stanów przejściowych i pośrednich dla wielu możliwych kombinacji katalizator–substrat. Z tych struktur wyodrębniają setki fizycznie znaczących deskryptorów, takich jak stopień zatłoczenia otoczenia katalizatora przy określonych atomach czy łatwość przemieszczania się elektronów. Liczby te są następnie wprowadzane do prostych modeli regresji liniowej, które łączą cechy strukturalne z mierzonymi wartościami selektywności.
Uczenie się z rzadkich danych dla prowadzenia nowych eksperymentów
Głównym osiągnięciem pracy jest maksymalne wykorzystanie rzadkich danych — ograniczonych kombinacji katalizatorów i substratów typowo raportowanych w artykułach naukowych. W jednym studium przypadku zespół analizuje reakcję niklową sprzęgającą epoksydy styrenowe z jodkami arylowymi. Pokazują, że deskryptory pochodzące z najbardziej odpowiedniego stanu przejściowego przewyższają te uzyskane z uproszczonych fragmentów katalizatora, mimo że bazowe obliczenia są tańsze. Dysponując tymi modelami, wirtualnie testują znacznie więcej ligandów dla istniejących par substratów i identyfikują nowe wybory katalizatorów, które podnoszą nadmiar enantomerowy w szczególnie opornych przypadkach, unikając przy tym dziesiątek niepotrzebnych eksperymentów.
Przenoszenie wiedzy między różnymi reakcjami
Podejście jest potężne, ponieważ można je przenieść na różne, lecz powiązane reakcje niklowe. W drugim zestawie badań autorzy łączą dane z kilku typów reakcji niklu, które wszystkie tworzą wiązania między atomami węgla hybrydyzowanymi sp3 a partnerami takimi jak grupy arylowe czy alkenylowe, nawet gdy dokładne warunki lub partnerzy sprzęgania się różnią. Budując modele na tych samych mechanistycznie znaczących deskryptorach, skutecznie przewidują enantioselektywność dla nowych ligandów, nowych kombinacji substratów, a nawet dla zupełnie nowej klasy reakcji tworzącej wiązania węgiel–węgiel, która nie była uwzględniona w zbiorze treningowym. Analiza, które deskryptory są najważniejsze, sugeruje także, który etap cyklu katalitycznego ustala ostateczną „ręczność” dla każdej rodziny reakcji.
Pomoc chemikom w szybszym rozpoczynaniu nowych reakcji
W końcowej demonstracji autorzy wykorzystują swój schemat deskryptorów razem z platformą optymalizacji bayesowskiej do zaprojektowania niklowo-katalizowanego sprzęgania acetali benzylicznych z jodkami arylowymi, które wcześniej nie były rozwijane asymetrycznie. Bazując na danych z literatury dotyczących innych reakcji, model rekomenduje małe zestawy obiecujących ligandów do przetestowania, szybko zbliżając się do najlepiej działającej klasy w zaledwie kilkudziesięciu eksperymentach. Dla chemika oznacza to praktyczne narzędzie do „zimnego startu” nowego projektu katalitycznego: wprowadzając garść wstępnych wyników, model może zasugerować, które ligandy chiralne najprawdopodobniej dostarczą wysoką enantioselektywność. Ogólnie badanie pokazuje, że starannie dobrane, niskokosztowe cechy obliczeniowe mogą przekształcić ograniczone dane z przeszłości w szeroko użyteczne wskazówki do budowy następnej generacji selektywnych reakcji.
Cytowanie: Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature 651, 637–646 (2026). https://doi.org/10.1038/s41586-026-10239-7
Słowa kluczowe: kataliza asymetryczna, sprzęganie krzyżowe niklowe, uczenie maszynowe w chemii, optymalizacja reakcji, predykcja enantioselektywności